

Incidencia nádorových ochorení je v našej populácii značne vysoká a neustále stúpa. Absolútna väčšina takýchto ochorení je sporadická, čiže náhodná. Existujú však rodiny, v ktorých sa špecifické typy onkologických ochorení opakujú, prípadne sa vyskytujú aj v nezvyčajne mladom veku. V týchto prípadoch môže v rodine ísť o genetickú predispozíciu. Približne v 5 – 10 % prípadov je táto predispozícia obyčajne veľmi silná a je viazaná na jeden konkrétny gén. Hovoríme, že ide o hereditárne (dedičné) typy nádorových ochorení. Často sú označované termínom hereditárne nádorové syndrómy, čím je vyjadrený polytropný charakter dedičnej onkologickej predispozície, ktorá sa s rozličnou mierou rizika môže vzťahovať aj na viacero orgánov alebo orgánových systémov. V nasledujúcom prehľade uvádzame najčastejšie typy hereditárnych onkologických syndrómov, s ktorými sa lekári rôznych medicínskych odborov môžu v praxi stretnúť.
Hereditárny karcinóm prsníka a ovárií (HBOC)
HBOC tvorí približne 10 % všetkých prípadov zhubného ochorenia prsníka. Najčastejšie ho spôsobujú mutácie génov BRCA1 a BRCA2. Približne v 10 – 20 % prípadov ide o ďalšie asociované gény TP53, RAD51, ATM, CHEK2, BRIP1, CDH1, STK11. U nosičiek mutácií uvedených génov vzrastá celoživotné riziko vzniku karcinómu prsníka na 65 – 85 %, čo je 8 – 10-násobné navýšenie všeobecného populačného rizika. Nádorové ochorenie sa objavuje už pred 40. rokom života, pričom v 60 % prípadov sa stretávame so sekundárnym výskytom karcinómu prsníka. Karcinóm ovária je častejší u nosičiek mutácie génu BRCA1 (60 %) než u nosičiek génu BRCA2 (10 – 20 %). Významne zvýšené je riziko nádorového postihnutia vajcovodov. Riziko postihnutia maternice je vzhľadom na populačné riziko navýšené 3 – 4-krát. S karcinómom prsníka sa u HBOC syndrómu stretávame často aj u mužov. Toto riziko je u nosičov mutácie BRCA2 zvýšené – oproti bežnej populácii – až stonásobne. Ďalšími tumormi asociovanými s nosičstvom mutácií génov BRCA1 alebo BRCA2 sú zhubné nádory prostaty, zažívacieho traktu, melanóm, prípadne aj nádory iného typu – v závislosti od pôsobenia rôznych rizikových faktorov. HBOC má autozómovo dominantnú dedičnosť.
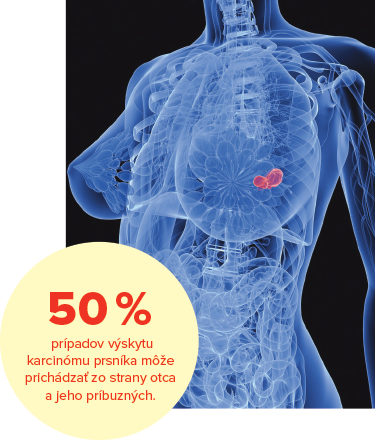
Indikácie na vyšetrenie BRCA génov
Familiárne formy:
- ak sa ochorenie vyskytlo u troch priamych príbuzných
- alebo u dvoch príbuzných, z ktorých je jeden vo veku do 50 rokov.
Nesmie sa pri tom zabúdať na príbuzných v otcovskej línii, keďže až v 50 % prípadov môže riziko karcinómu prsníka prichádzať zo strany otca a jeho príbuzných.
Sporadické formy:
- včasný výskyt ochorenia u pacientky pred 40. rokom života,
- obojstranná forma karcinómu prsníka, prípadne kombinácia rakoviny prsníka a vaječníka u tej istej osoby,
- výskyt karcinómu prsníka u muža,
- špecifické typy karcinómov (medulárny typ, negativita hormonálnych receptorov a HER2) u osôb mladších než 50 rokov.
Zdraví členovia rodiny sa testujú až po identifikácii patologickej mutácie u symptomatického príbuzného. Ako prví v rodokmeni sa testujú len veľmi zriedkavo, keďže výpovedná hodnota takéhoto vyšetrenia je značne obmedzená. Prediktívne testovanie sa robí až po dosiahnutí plnoletosti.
Prevencia odporúčaná ženám – nosičkám mutácie génu BRCA1/2
Preventívne vyšetrenia prsníkov:
- samovyšetrovanie prsníkov raz mesačne po ukončení menštruácie,
- klinické vyšetrenie prsníkov odborným lekárom dvakrát ročne,
- USG prsníkov raz ročne od 25 rokov,
- MG raz ročne od 30 rokov,
- NMR prsníkov raz ročne od 25 rokov.
Dôkladné pravidelné samovyšetrovanie prsníkov, fyzikálne vyšetrenie prsníkov skúseným klinickým lekárom a vyšetrenie prsníkov zobrazovacími metódami je prepojená triáda, v ktorej má každá zložka svoj nenahraditeľný význam. 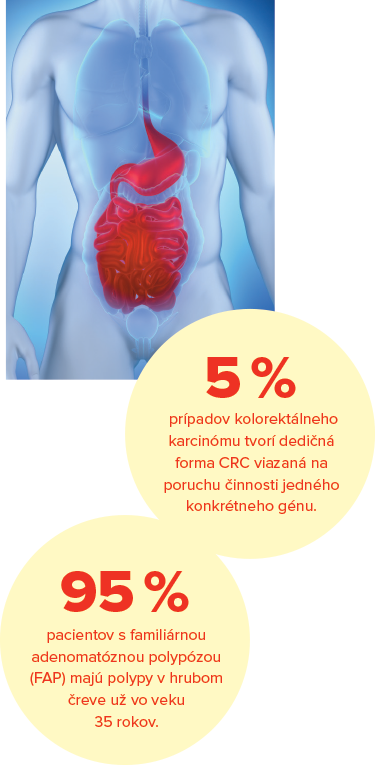
Preventívne vyšetrenia vaječníkov:
- gynekologické vyšetrenie a transvaginálny ultrazvuk dvakrát ročne od 21 rokov,
- vyšetrenie nádorového markera CA 125 raz ročne.
Preventívne vyšetrenia zamerané na iné typy nádorových ochorení:
- emokult, kolonoskopia, kožné vyšetrenie, abdominálna USG.
Nositeľky patologických mutácií génov BRCA1/2 treba informovať aj o možnosti profylaktických chirurgických zákrokov, ktorými sú bilaterálna adnexektómia a bilaterálna mastektómia.
Prevencia odporúčaná mužom – nosičom mutácie génu BRCA1/2:
- samovyšetrovanie prsníkov a semenníkov raz mesačne,
- USG prsníkov raz ročne od veku 30 rokov,
- urologické vyšetrenie aj s PSA markerom raz ročne od veku 45 rokov,
- ďalšie vyšetrenia ako vyšetrenie stolice na OK, kolonoskopia, abdominálna USG, kožné vyšetrenie.
V prípade preukázania nosičstva patologickej mutácie iného asociovaného génu zodpovedajú preventívne opatrenia príslušným odporúčaniam pre tieto gény. Ak sa pri splnení indikačných kritérií patologická mutácia u pacientky s karcinómom prsníka nepreukáže, pre jej príbuzné je vypočítavané tzv. empirické riziko vzniku karcinómu prsníka podľa Clausových tabuliek, na základe ktorého sa vypracuje odporúčaný dispenzarizačný plán.
Hereditárny nepolypózny kolorektálny karcinóm (HNPCC) = Lynchov syndróm (LS)
Incidencia kolorektálneho karcinómu (CRC) je u nás jedna z najvyšších na svete. Asi 5 % prípadov tvorí dedičná forma CRC viazaná na poruchu činnosti jedného konkrétneho génu. Zodpovedné gény sú tzv. MMR gény – MLH1, MSH2, MSH6, PMS2. Dedičnosť je autozómovo dominantná. Najčastejšou formou je HNPCC, ktorá sa spája s 20 – 80-percentným rizikom vzniku karcinómu hrubého čreva a 25 – 70-percentným rizikom vzniku karcinómu endometria u žien. Kolorektálny karcinóm sa môže s vysokou pravdepodobnosťou aj zopakovať. V hrubom čreve sa niekedy vyskytuje menšie množstvo polypov. Ďalšími asociovanými tumormi s HNPCC sú karcinóm vaječníkov (3 – 13 %), žalúdka (2 – 13 %), tenkého čreva (4 – 7 %), močových ciest (1 – 12 %), hepatobiliárneho systému (2 %) a nádory mozgu (1 – 4 %). Niektoré štatistiky uvádzajú aj zvýšené riziko karcinómu prsníka v takýchto rodinách. Nádorové ochorenia sa nezriedka objavujú už v mladom veku a bývajú často viacpočetné. Oproti iným typom karcinómov hrubého čreva majú nádory špecifický biologický charakter vyznačujúci sa okrem iného rýchlym vývojom (1 – 3 roky) a relatívne lepšou prognózou.
Indikácie na vyšetrenie MMR génov
Pôvodné, tzv. Amsterdamské kritériá – známe aj ako 3-2-1 kritériá – zohľadňovali predovšetkým familiárny výskyt CRC a asociovaných tumorov. V súčasnosti sú najviac akceptované revidované kritériá z Bethesdy, ktoré zahŕňajú aj sporadické formy CRC:
- výskyt ochorenia u troch priamych príbuzných,
- alebo u dvoch priamych príbuzných, z ktorých je jeden vo veku do 50 rokov,
- včasný výskyt CRC do 50. roku života,
- viacpočetný výskyt CRC alebo asociovaného tumoru u jedného pacienta,
- CRC s histologickým nálezom zodpovedajúcim vysokému stupňu MSI u pacienta do 60 rokov (medulárny rast, mucinózny charakter často s prítomnosťou prstencových buniek, tumor infiltrujúce lymfocyty, pseudozápalová lymfocytárna infiltrácia).
HNPCC je geneticky heterogénnou klinickou jednotkou, preto má štandardný vyšetrovací harmonogram určitú postupnosť. Genetická analýza sa začína skríningovým vyšetrením nádorového tkaniva na prítomnosť mikrosatelitovej instability (MSI) a imunohistochemickým vyšetrením nukleárnej expresie proteínových produktov príslušných génov (ICH). V prípade zistenia pozitívneho nálezu sa pokračuje ďalej DNA analýzou príslušného génu z krvi pacienta. Prediktívne testovanie sa pri známej patologickej mutácii v rodine robí až po dosiahnutí plnoletosti.
Odporúčaná prevencia:
- kolonoskopické vyšetrenie každé 1 – 2 roky od 20 rokov,
- vyšetrenie stolice na OK v medziobdobí,
- gastroskopia raz za 3 – 4 roky od 35 rokov,
- gynekologické vyšetrenie vrátane TVS a onkomarkera CA 125 raz ročne od 30 rokov,
- spiračná biopsia endometria raz ročne od 30 rokov,
- vyšetrenie moču a urologické vyšetrenie od 40 rokov.
Z ďalších vyšetrení sú to vyšetrenia prsníkov, onkodermatologické vyšetrenie a orientačné neurologické vyšetrenie. Ženy – nosičky patologických mutácií musia byť informované aj o možnosti preventívnej hysterektómie po skončení reprodukčných povinností.
Hereditárne polypózne syndrómy
Ide o skupinu viacerých dedičných syndrómov. Dnes už u väčšiny z nich poznáme genetický podklad a vieme vyšetriť zodpovedné gény.
1. Familiárna adenomatózna polypóza (FAP)
Klasická forma familiárnej adenomatóznej polypózy asociovaná s génom APC je charakterizovaná výskytom viac než stovky, často až tisícov, adenomatóznych polypov v hrubom čreve. Typ dedičnosti je autozómovo dominantný. Polypy sa začínajú tvoriť v 2. – 4. decéniu, pričom vo veku 35 rokov má polypy 95 % osôb. Ochorenie je spojené s vysokým rizikom vzniku kolorektálneho karcinómu, často už v mladom veku. Polypóza sa môže spájať aj s inými extrakolonickými príznakmi vrátane hypertrofie pigmentového epitelu sietnice, osteómami, poruchami dentinogenézy, desmoidmi, epidermoidnými cystami a adenómami vyšších častí GIT-u. FAP sa okrem prakticky 100-percentného rizika vývoja CRC vyznačuje aj zvýšeným rizikom vzniku meduloblastómu, papilárneho karcinómu štítnej žľazy, hepatoblastómu, karcinómu žalúdka a pankreasu. Okrem klasickej formy FAP sa môžeme stretnúť aj s atenuovanou formou, pri ktorej je množstvo polypov obvykle menej než 100 a vyskytujú sa spravidla vo vyššom veku. Závažnosť polypózy a riziko vývoja extrakolonických príznakov a malignít sa dá odhadnúť aj na základe fenotypovo-genotypovej korelácie, t. z. podľa lokalizácie zistenej patologickej mutácie v rámci génu APC a jej rozsahu.
MUTYH asociovaná polypóza (MAP)
Miernejšia forma adenomatóznej polypózy (v počte polypov 3 – 100) a často so sporadickým výskytom v rodinách. Môže byť asociovaná s bialelickým výskytom patologických mutácií v géne MUTYH. Ide o zriedkavý autozómovo recesívny typ polypózy, na ktorú je potrebné myslieť v prípade negatívneho výsledku vyšetrenia génu APC.
Indikácie na vyšetrenie génov APC a MUTYH:
- všetky formy difúznej adenomatóznej polypózy kolorekta.
Prediktívne testovanie sa vzhľadom na možnosť výskytu závažných prejavov ochorenia už vo včasných vekových kategóriách robí v akomkoľvek veku.
Odporúčaná prevencia:
Sledovanie a preventívne zákroky by u pacientov s potvrdenou patologickou mutáciou mali byť individualizované, s prihliadnutím na genotyp a fenotyp pacienta.
Klasická forma FAP si vyžaduje:
- pravidelné sigmoidoskopické alebo kolonoskopické vyšetrenie od veku 10 – 12 rokov v 1 – 2-ročných intervaloch do záchytu polypózy,
- po manifestácii polypózy kolonoskopické vyšetrenie v 1-ročných intervaloch, pri zistení ťažkého stupňa polypózy alebo ťažkej dysplázie je odporúčaná totálna kolektómia s kontrolami ileo-análneho pouchu v 2-ročných intervaloch alebo proktokolektómia s kontrolami zostatku rekta v 6-mesačných intervaloch,
- gastroduodenoskopia; podľa genotypu každé 1 – 3 roky od 25 rokov,
- skríning hepatoblastómu v detskom veku do 5 rokov,
- vyšetrenie štítnej žľazy v ročných intervaloch.
Užívanie nesteroidných antiflogistík (NSAID) podľa sledovaní napomáha regresii adenómov a redukuje ich počet hlavne u pacientov po proktokolektómii. Desmoidné tumory si vzhľadom na vysoký stupeň rekurencie vyžadujú opatrný chirurgický prístup, uprednostňuje sa nesteroidná antiinflamačná liečba, antiestrogénová terapia, chemoterapia alebo rádioterapia na špecializovaných pracoviskách. Pacienti s atenuovanou formou FAP a s MUTYH asociovanou adenomatóznou polypózou vyžadujú individuálne, menej frekventované sledovanie od veku 21 – 25 rokov.
2. Peutzov-Jeghersov syndróm (PJS)
Ide o menej častý polypózny syndróm charakteristický nápadnými mukokutánnymi pigmentáciami v okolí úst, nozdríl, na bukálnej sliznici a v perianálnej oblasti. Pigmentácie sú často pozorované už v prvom roku života a ich intenzita sa vekom stráca. Syndróm sa dedí autozómovo dominantne. Zodpovedným génom je STK11. Vedúcim symptómom u PJS sú hamartomatózne polypy, hlavne v jejune, ileu, menej v hrubom čreve, žalúdku a duodene, ktoré sú častou príčinou invaginácií a príznakov ilea už vo včasnom veku. V hrubom čreve sa môžu vyskytovať aj iné typy polypov vykazujúcich adenomatózne zmeny. Mutácie génu STK11 spôsobujú zvýšené riziko rôznych typov onkologických ochorení s kumulatívnym celoživotným rizikom do 80 %, z ktorých sú najčastejšie nádory zažívacieho traktu (66 %), karcinóm prsníka (32 %), gonadálne tumory ovárií, vajcovodov, krčka maternice a semenníkov u mužov.
Indikácie na vyšetrenie génu STK11:
- 2 alebo viac hamartomatóznych polypov v GIT-e,
- jeden hamartomatózny polyp a mukokutánne pigmentácie,
- jeden hamartomatózny polyp a pozitívna rodinná anamnéza na PJS.
Prediktívne testovanie sa robí u rizikových členov rodiny aj v detskom veku.
Odporúčaná prevencia:
- gastroduodenoskopické a kolonoskopické vyšetrenie každé 2 roky od 8 rokov,
- kapsulárna endoskopia tenkého čreva každé 2 roky od 8 rokov,
- abdominálna USG, endoskopický ultrazvuk a vyšetrenie onkomarkera CA 19-9 pre riziko karcinómu pankreasu každé 1 – 2 roky od 25 rokov,
- prsníková prevencia a gynekologická prevencia u žien pravidelne od 18 rokov, treba zvážiť aj preventívnu hysterektómiu a adnexektómiu po 35. roku života,
- kontroly testes u mužov od 20 rokov.
3. Juvenilný polypózny syndróm (JPS)
Ochorenie je charakteristické výskytom mnohopočetných juvenilných (hamartomatóznych) polypov v priebehu celého GIT-u, predovšetkým v hrubom čreve a konečníku, menej často v žalúdku a tenkom čreve. Výskyt polypov zaznamenávame už v detskom veku, pričom najťažšia generalizovaná forma JPS sa môže vyvinúť už okolo 2. roku života a má značne nepriaznivú prognózu. JPS sa spája aj so zvýšeným rizikom výskytu zhubných nádorov zažívacieho traktu a pankreasu s celoživotným kumulatívnym rizikom odhadovaným na 68 %. Dedičnosť je autozómovo dominantná. Zodpovedné gény sú SMAD4 a BMPR1A.
Indikácie na vyšetrenie génov SMAD4 a BMPR1A:
- viac ako 5 juvenilných polypov v kolorekte alebo celom GIT-e,
- akýkoľvek počet juvenilných polypov a pozitívna rodinná anamnéza JPS.
Zárodočné mutácie uvedených génov sa zisťujú približne u 50 – 60 % pacientov s JPS. Prediktívne sa testujú rizikoví členovia rodiny už od včasného veku.
Odporúčaná prevencia:
- kolonoskopické vyšetrenie v 1 – 3-ročných intervaloch od 10 – 15 rokov, prípadne od prvých symptómov,
- gastroduodenoskopia v 1 – 3-ročných intervaloch od 10 – 15 rokov,
- zobrazovacie vyšetrenia zamerané na tenké črevo a pankreas,
- sledovanie krvného obrazu a u patologických mutácií génu SMAD4 aj možných cievnych malformácií (RTG hrudníka, MR mozgu, USG pečene).
V prípade závažnej polypózy a familiárneho výskytu CRC sa zvažuje možnosť kolektómie.
4. Cowdenov syndróm
Cowdenov syndróm, najčastejší zo skupiny PTEN hamartoma tumor syndrómov (PTHS), je charakteristický výskytom mnohopočetných hamartómov vrátane hamartomatóznych polypov hrubého čreva, hamartómov prsníkov a u časti pacientov aj rôznorodých kožných a slizničných lézií. Asociovaný je aj so zvýšeným rizikom benígnych, ale aj malígnych nádorov štítnej žľazy (10 %), prsníka (25 – 50 %), endometria a iných orgánov. Príznaky sa manifestujú obyčajne v 2. – 3. dekáde života, pričom expresia príznakov môže byť značne variabilná. To ho pred manifestáciou extrakolonických znakov niekedy ťažko odlišuje od PJS alebo JPS. Zvýšené riziko CRC však u pacientov s Cowdenovým syndrómom nebolo zaznamenané. Syndróm sa dedí autozómovo dominantne, zodpovedným génom je PTEN.
Indikácie na vyšetrenie génu PTEN:
Klinická diagnóza Cowdenovho syndrómu je postavená na prítomnosti:
- typických mukokutánnych lézií (trichilemony tváre, akrálna keratóza, papilomatózne lézie, slizničné lézie),
- veľkých kritérií (gangliocytóm mozočku, karcinóm prsníka, karcinóm štítnej žľazy, karcinóm endometria, prítomnosť makrocefálie),
- malých kritérií (hamartomatózne intestinálne polypy, adenómy štítnej žľazy, lipómy, fibrómy, karcinóm obličky, prsníková fibróza).
Odporúčaná prevencia:
- kožné vyšetrenie, vyšetrenie moču, USG brušných orgánov, USG štítnej žľazy raz ročne od 20 – 25 rokov,
- kolonoskopia od 50 rokov v 5-ročných intervaloch a pravidelné vyšetrenie stolice na OK,
- raz ročne prsníková a gynekologická prevencia u žien od 25 rokov, od 35 rokov raz ročne MRI prsníkov,
MG a aspiračná biopsia endometria.
Li-Fraumeniho syndróm (LFS)
Ide o závažnú geneticky podmienenú predispozíciu pre vznik rôznorodých onkologických ochorení, ktoré sa manifestujú už v nezvyčajne mladom veku. Celoživotné riziko vzniku onkologického ochorenia je pre nositeľov závažného typu mutácie génu TP53 73 – 100 %, v závislosti od pohlavia. 77 % všetkých malignít u pacientov s LFS tvoria karcinóm prsníka, sarkómy mäkkých tkanív, osteosarkóm, nádory kôry nadobličiek, leukémie a nádory mozgu. So zvýšenou mierou rizika sa objavujú aj ďalšie malignity ako CRC, karcinóm pankreasu, karcinóm žalúdka, germinálne nádory a malígny melanóm. 45 % nádorov sa diagnostikuje do veku 45 rokov. LFS sa dedí autozómovo dominantne, zodpovedným génom je TP53.
Indikácia na vyšetrenie TP53 génu:
Klasické kritériá pre diagnózu LFS sú:
- proband so sarkómom diagnostikovaným do veku 45 rokov a súčasne jeden príbuzný 1. st. s akýmkoľvek nádorom do veku 45 rokov a ďalší príbuzný 1. alebo 2. st. s akýmkoľvek nádorom do veku 45 rokov alebo sarkómom bez ohľadu na vek.
Modifikované kritériá pre LFS podľa Chompretovej zahŕňajú:
- proband s nádorom detského veku, sarkómom, tumorom mozgu alebo adrenálnym kortikálnym nádorom diagnostikovaný do 46. roku a zároveň príbuzný 1. alebo 2. st. s nádorom typickým pre LFS do 56. roku života,
- proband s mnohopočetnými nádormi, z ktorých sú dva typické pre LFS a jeden vznikol do veku 46 rokov,
- pacient s adrenokortikálnym karcinómom,
- pacientka s karcinómom prsníka do 36 rokov bez známej mutácie génov BRCA1/2.
Pravdepodobnosť záchytu patologickej mutácie je v rodinách spĺňajúcich klasické kritériá asi 50 %, v rodinách spĺňajúcich modifikované kritériá podľa Chompretovej asi 20 %. Prediktívne testovanie sa robí v dospelom veku, avšak vzhľadom na možnosť výskytu nádorového ochorenia treba preventívne sledovať všetky deti v rodine. Prediktívne testovanie v detskom veku je indikované po zvážení prínosu vyšetrenia v špecifických situáciách.
Genetické testovanie predispozície na širokú škálu onkologických ochorení asociovaných s LFS sa robí po dôkladnej analýze osobnej a rodinnej situácie probanda, pričom sa musí brať do úvahy značná heterogenita LFS asociovaných nádorov a psychologický dopad pozitívneho výsledku testovania na pacienta a jeho rodinu.
Odporúčaná prevencia:
Deti majú raz ročne indikované základné fyzikálne vyšetrenie, KO, vyšetrenie moču, abdominálnu USG, neurologické vyšetrenie, očné vyšetrenie, vyžaduje sa informovanosť pediatra aj rodiny o možnosti objavenia sa nešpecifických príznakov a o potrebe kontaktovať v takýchto situáciách detského onkológa.
Dospelí:
- fyzikálne vyšetrenie, KO a moč raz ročne,
- abdominálna USG raz ročne,
- kolonoskopické vyšetrenie raz za 2 – 3 roky od 30 rokov,
- a gastroduodenoskopické vyšetrenie raz za 3 roky od 35 rokov,
- neurologické vyšetrenie a MRI mozgu raz ročne,
- vyšetrenie prsníkov a USG dvakrát ročne, MRI prsníkov raz ročne,
- gynekologické vyšetrenie s TVS dvakrát ročne,
- u mužov vyšetrovanie semenníkov a prostaty.
Všeobecne sa treba vyhýbať zobrazovacím vyšetreniam na báze ionizujúceho žiarenia vzhľadom na zvýšenú rádiosenzitivitu nosičov patologickej mutácie génu TP53.
Von Hippelova-Lindauova choroba (VHL)
VHL sa vyznačuje tvorbou viacpočetných hemangioblastómov, najčastejšie v mozočku, mozgovom kmeni, mieche a v sietnici (60 – 80 % pacientov), pankreatických cýst a cystadenómov (20 – 50 %), epididymálnych cýst (7 – 27 %) a nádorov endolymfatického vaku vedúcich k strate sluchu. Hemangiómy a cysty sa môžu tvoriť aj na iných orgánoch a tkanivách. Zvýšené riziko vzniku nádorového ochorenia sa u VHL týka hlavne karcinómu obličky (40 %), ktorý sa môže vyskytovať v solitárnej ale aj mnohopočetnej a bilaterálnej forme. Ďalšou formou nádorového ochorenia sú neuroendokrinné tumory, najčastejšie feochromocytóm nadobličky, ale aj v iných lokalizáciách. Menej často to bývajú pankreatické neuroendokrinné tumory, ktoré sa však môžu správať ako zhubné nádory s tvorbou metastáz. VHL sa dedi autozómovo dominantne, zodpovedným génom je VHL.
Indikácie na vyšetrenie génu VHL:
- viacpočetné hemangioblastómy (CNS, sietnica),
- hemangioblastóm (CNS, sietnica) a súčasne výskyt renálnych alebo pankreatických cýst, feochromocytómu alebo karcinómu obličky,
- jeden z vyššie uvedených príznakov a pozitívna rodinná anamnéza na VHL.
Prediktívne testovanie sa robí už od detského veku vzhľadom na možnosť manifestácie závažných príznakov vo včasnom veku. Ochorenie má temer kompletnú penetranciu do veku 65 rokov.
Odporúčaná prevencia:
V závislosti od veku:
- očné vyšetrenie raz ročne,
- fyzikálne a neurologické vyšetrenie raz ročne,
- audiologické vyšetrenie raz za 2 roky,
- MRI mozgu a miechy raz za 1 – 2 roky,
- abdominálna USG a MRI obličiek, nadobličiek a pankreasu raz ročne a CT brucha raz za 3 roky,
- abdominálna USG a MRI obličiek, nadobličiek a pankreasu raz ročne a CT brucha raz za 3 roky,
V prípade manifestácie konkrétneho príznaku treba harmonogram vyšetrení cielene modifikovať.
Mnohopočetná endokrinná neoplázia typu 2 (MEN2 syndróm)
MEN2 syndróm je ochorenie sprevádzané súčasným alebo postupným výskytom medulárneho karcinómu štítnej žľazy, unilaterálneho alebo bilaterálneho feochromocytómu a primárnej hyperparatyreózy (MEN2A) alebo marfanoidným habitom, feochromocytómom a radom ďalších lézií ako neurinómy a gangliómy hlavne zažívacieho traktu (MEN2B). Ide o autozómovo dominantný typ dedičnosti.
Medulárny karcinóm štítnej žľazy (MTC) je nádor vychádzajúci z parafolikulárnych buniek (C-buniek) štítnej žľazy produkujúcich kalcitonín a predstavuje asi 4 – 10 % všetkých nádorov štítnej žľazy. Ide o agresívnu formu nádorového ochorenia s rýchlou tvorbou metastáz. Asi 25 % MTC sa vyskytuje ako familiárna forma s dominantným typom dedičnosti. Môže sa vyskytovať ako samostatná forma familiárneho MTC (FMTC) alebo v rámci MEN2 syndrómu spolu s feochromocytómom a hyperparatyreózou. Plný rozvoj ochorenia nastáva priemerne v 3. – 4. decéniu. Forma MEN2B je agresívnejšia forma, manifestuje sa už v detskom veku. Táto forma je rozpoznateľná aj na základe typickej somatickej symptomatológie. Dedičnosť je autozómovo dominantná, zodpovedným génom je protoonkogén RET.
Indikácie na vyšetrenie protoonkogénu RET:
Na vyšetrenie sú indikovaní všetci pacienti s MTC a pacienti s feochromocytómom predovšetkým v mladom veku a s bilaterálnym alebo multifokálnym výskytom. Vyšetrenie je vzhľadom na zvýšené riziko MTC odporúčané aj pacientom s Hirschsprungovou chorobou.
Prediktívne testovanie rizikových členov rodiny sa robí v každom veku, u detí je to čím skôr po narodení. U MEN2 syndrómu sú známe fenotypovo-genotypové korelácie patologických mutácií, čiže na základe lokalizácie a typu mutácie vieme odhadnúť jej fenotypový dopad a agresivitu.
Odporúčaná prevencia:
Profylaktická tyreoidektómia sa vykonáva už v detskom veku a to podľa zaradenia patologickej mutácie do rizikovej skupiny. U najrizikovejších foriem je odporúčaná už v prvých mesiacoch života. Pacienti aj po TTE by z dôvodu rozvoja reziduálnej formy MTC mali byť sledovaní pravidelným vyšetrením bazálnej aj stimulovanej hladiny kalcitonínu. Podľa zisteného typu mutácie sa odporúča ďalšie sledovanie krvného tlaku, hladín plazmatických alebo močových katecholamínov a kalcia, fosforu a parathormónu.
Syndróm familiárneho malígneho melanómu (FAMMM)
Asi 10 % malígnych melanómov sa vyskytuje familiárne. V niektorých takýchto rodinách sa objavujú mnohopočetné névy – hlavne na trupe a končatinách – vykazujúce väčšinou atypie melanocytov s lymfocytárnou infiltráciou a fibropláziou. V iných rodinách však takéto kožné prekanzerózne lézie nezaznamenávame. Asi 20 – 40 % familiárnych melanómov má identifikovanú patologickú mutáciu v géne CDKN2A. Nosičstvo takejto predispozície zvyšuje celoživotné riziko vzniku malígneho melanómu na 58 – 92 %, čo je 30 – 70-násobne vyššie oproti všeobecnému populačnému riziku. Okrem rizika malígneho melanómu majú nosiči patologickej mutácie zvýšené riziko karcinómu pankreasu (20 %) a karcinómu prsnej žľazy. Asociácia s inými typmi nádorových ochorení zatiaľ nebola jednoznačne preukázaná (tumory mozgu, zažívacieho traktu). Ochorenie má autozómovo dominantnú dedičnosť, zodpovedným génom je CDKN2A, ojedinele CDK4.
Indikácie na vyšetrenie génu CDKN2A
Familiárny výskyt:
- dva malígne melanómy u príbuzných 1. alebo 2. st., z toho jeden pred 50. rokom života,
- tri a viac malígnych melanómov v rodine,
- dva malígne melanómy a karcinóm pankreasu alebo prsníka v rodine.
Sporadický výskyt:
- opakovaný výskyt malígneho melanómu u pacienta, prvý prípad pred 50. rokom života,
- viacpočetná malignita u pacienta, t. z. malígny melanóm/pankreas/prsník, z toho prvý prípad do 50. roku života.
Prediktívne testovanie sa robí aj v detstve.
Odporúčaná prevencia:
- mesačné samovyšetrovanie pokožky,
- onkodermatologické vyšetrenie 2-krát do roka s včasným chirurgickým odstránením rizikových znamienok,
- očné vyšetrenie raz ročne,
- abdominálna USG raz ročne od 30 rokov, podľa rodinnej anamnézy aj ďalšie vyšetrenia na pankreas,
- prsníková prevencia raz ročne,
- kolonoskopické vyšetrenie od veku 45 rokov v 5 – 10-ročných ročných intervaloch.
V rizikových rodinách je veľmi dôležitá primárna prevencia spočívajúca v ochrane pred UV žiarením a vo vylúčení fajčenia.
Hereditárny difúzny karcinóm žalúdka (HDGC)
HDGC predstavuje závažnú genetickú predispozíciu pre vznik difúzneho karcinómu žalúdka. Ide o typ málo diferencovaného adenokarcinómu, pri ktorom nádorové bunky difúzne rastú a infiltrujú žalúdočnú stenu bez toho, aby tvorili solídnu nádorovú masu, pričom sliznica žalúdka nevykazuje makroskopické ani mikroskopické zmeny. Malígne bunky majú charakteristický vzhľad buniek pečatného prsteňa, čo je spôsobené zvýšenou akumuláciou intracelulárneho mucínu. Priemerný vek manifestácie ochorenia je 38 rokov, pričom výskyt je zaznamenaný už aj v 2. decéniu. Kumulatívne riziko vzniku difúzneho karcinómu žalúdka u nosičov patologickej mutácie je 67 – 83 %, ženy majú vyššie riziko. Včasná diagnostika je vzhľadom na difúzny podslizničný rast problematická, väčšina pacientov má pokročilý stupeň ochorenia už v dobe stanovenia diagnózy. Dedičnosť HGDC je autozómovo dominantná. Zodpovedným génom je CDH1. Nosiči mutácie CDH1 génu majú zvýšené riziko aj iných malignít, a to lobulárneho karcinómu prsníka (40 – 50 %) a kolorektálneho karcinómu s výraznou intracelulárnou mukoprodukciou (karcinóm z buniek pečatného prsteňa).
Indikácie na vyšetrenie génu CDH1:
- traja príbuzní 1. alebo 2. st. s karcinómom žalúdka (aspoň jeden difúzneho typu),
- dvaja príbuzní 1. alebo 2. st. s karcinómom žalúdka (z toho jeden difúzneho typu do 50 rokov),
- sporadický výskyt difúzneho karcinómu žalúdka u pacienta do 40 rokov,
- duplexný výskyt difúzneho karcinómu žalúdka a lobulárneho karcinómu prsníka alebo CRC z pečatných buniek (aspoň jeden do 50 rokov),
- viacpočetný lobulárny karcinóm prsníka u jednej pacientky vo veku do 50 rokov.
Viac ako 50 % prípadov HDGC nemá mutáciu génu CDH1. Uvažuje sa o genetickej heterogenite ochorenia s účasťou iných – zatiaľ neznámych – génov. Vzhľadom na neúplnú penetranciu génu sa prediktívne testovanie robí aj u starších členov rodiny. Prediktívne testovanie detí a mladistvých do 18 rokov sa robí len vo výnimočných situáciách závažného familiárneho výskytu ochorenia v mladom veku.
Odporúčaná prevencia:
Metódou voľby u pozitívne testovaných jedincov je profylaktická kompletná gastrektómia, ktorá je jediným spoľahlivým profylaktickým opatrením. Všetky ďalšie vyšetrenia vrátane gastroskopie s početnými biopsiami v polročných intervaloch – a to aj s využitím chromoendoskopie, CT žalúdka, prípadne PET scanu – nezabezpečujú dostatočne včasný záchyt ochorenia. To potvrdzujú aj pozitívne histologické nálezy u pacientov po preventívnej gastrektómii, u ktorých boli výsledky skríningových testov negatívne.
Z ďalších preventívnych vyšetrení je to mamologická prevencia od veku 35 rokov v polročných intervaloch s využitím USG a MRI prsníkov a kolonoskopické vyšetrenia od veku 40 rokov v 3 – 5-ročných intervaloch.
Záver
V boji proti väčšine zhubných nádorov zatiaľ nepoznáme účinnú primárnu prevenciu, ktorá by zabraňovala vzniku týchto ochorení. Preto je jednoznačnou prioritou včasný záchyt, ktorý pri neustálom zvyšovaní efektivity liečby umožňuje znížiť úmrtnosť na nádorové ochorenia a zlepšiť kvalitu života pacientov. Odkrývanie dedičných predispozícií a genetické testovanie rizikových osôb sú v tejto oblasti nespochybniteľným prínosom. V rámci rodiny je tak možné odhaliť rizikových členov a zahájiť u nich účinné preventívne opatrenia často vo veľmi mladom veku, t. j. v dobe, keď sa na takéto ochorenia väčšinou ešte nemyslí.
Úlohou lekárov prvého kontaktu a príslušných lekárov – špecialistov je vytipovať v populácii rizikovú skupinu pacientov a odoslať ich na špecializované genetické vyšetrenie. Všetky genetické testy indikuje klinický genetik po dôkladnom oboznámení pacienta s možnosťami, ale aj úskaliami genetického testovania a interpretácie jeho výsledkov. Po odhalení genetickej predispozície je úlohou príslušných odborníkov zabezpečiť a dôsledne dodržiavať genetikom vypracovaný program preventívnych opatrení, ktorý dáva týmto pacientom lepšiu šancu na prežitie, a tým aj lepšie vyhliadky do budúcnosti.
Molekulárno-genetická diagnostika všetkých uvedených ochorení je na Slovensku dostupná. Kontakt na príslušné ambulancie klinickej genetiky je uvedený na webovej stránke Slovenskej spoločnosti lekárskej genetiky.




