

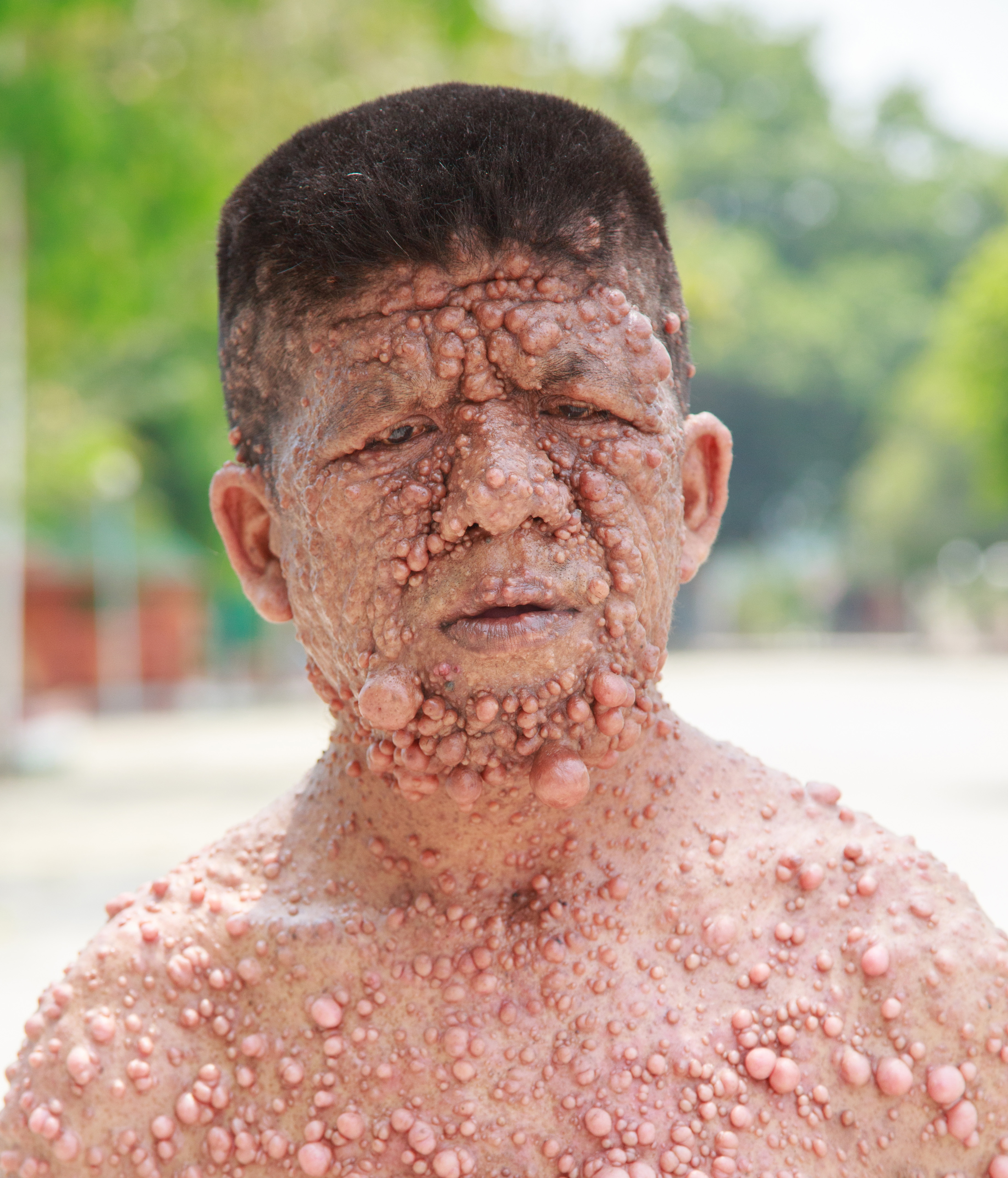
Aj keď k rozlúsknutiu genetickej podstaty neurofibromatózy na úrovni DNA došlo koncom minulého tisícročia, toto ochorenie bolo prvýkrát zobrazené na maľbách už v 13. storočí. Roku 1882 ho Frederick von Recklinghausen popísal ako prvý. Avšak vďaka Josephovi Merrickovi, ktorý sa na pódiách amatérskych divadiel Anglicka 19. storočia preslávil ako Sloní muž, sa neurofibromatóza stala morbídnou atrakciou pre davy zvedavcov.
Neurofibromatóza je geneticky podmienené neurokutánne ochorenie, najčastejšie autozomálne dominantné ochorenie vôbec. Rozlišujeme neurofibromatózu typu 1 (NF1), ktorá tvorí okolo 90 % prípadov NF, neurofibromatózu typu 2 (NF2) a schwannomatózu. Vďaka typickému kožnému nálezu NF1 prichádzajú dospelí pacienti od špecialistov, ba aj praktických lekárov do ambulancie klinického genetika takmer s istou diagnózou. Nejde len o nepríjemný estetický dojem. Málokto si uvedomuje predispozíciu k vzniku malígnych nádorov a k rastu enormne objemných benígnych tumorov u pacientov.
Klasické príznaky NF1
Klinické prejavy NF1 sú extrémne pestré. Aj v rámci jednej rodiny sa vyskytuje značná intrafamiliárna variabilita príznakov. Viac než 40 % pacientov s NF1 má už pri narodení prvé príznaky ochorenia, do konca 1. roka sa toto číslo zvyšuje na 60 %. Ako prvý príznak nastupujú väčšinou ostro ohraničené, oválne alebo okrúhle, homogénne sa farbiace škvrny bielej kávy (ďalej len „café-au-lait“) v niveau kože. Nevyskytujú sa palmárne ani plantárne. Postupne pribúdajú drobné pigmentové ložiská pripomínajúce pehy, tzv. freckling v oblasti axíl a inguín. Lischove noduly (hamartómy dúhovky) sa objavujú v ranom detstve a sú prítomné takmer u všetkých dospelých postihnutých. Diagnostikuje sa spoľahlivo vyšetrením štrbinovou lampou. Gliómy CNS sú veľmi časté, obzvlášť glióm optickej dráhy, mozgového kmeňa a mozočka. Glióm n. opticus sa vyvinie do 6. roku, neskôr už nedôjde k jeho vzniku. Môže byť klinicky nemý alebo symptomatický a zapríčiniť poruchu farbocitu, zorného poľa, zníženú kvalitu zraku, strabismus, edém papily až slepotu. Veľakrát dôjde k spontánnej regresii gliómu. Obojstranný glióm n. opticus je prítomný takmer iba u pacientov s NF1. Glióm n. opticus sa správa oveľa benígnejšie, keď je súčasťou syndrómu NF1 ako izolovaný nález. Neurofibrómy, podľa ktorých je pomenovaná choroba, sa objavujú od puberty. Ich počet a rozmer postupne narastá počas života. Môžu svrbieť a bolieť. V období gravidity nastáva rýchly vznik nových neurofibrómov. Plexiformný neurofibróm, na rozdiel od kutánneho alebo subkutánneho neurofibrómu, je obyčajne kongenitálny a postihuje nervové korene a splete. Preniká aj do okolitých tkanivových štruktúr. Môže byť detegovateľný hneď pod povrchom tela, ale jeho rozsah je bez MRI vyšetrenia ťažko odhadnuteľný. Na prítomnosť hlboko uloženého plexiformného neurofibrómu môže upozorniť hyperpigmentovaná alebo drsnejšia koža nad samotnou léziou. Veľakrát ostáva asymptomatický. Zvykne narásť počas puberty. Kostné anomálie sú prítomné už pri narodení, ale k ich prejavom dochádza niekedy až počas dospievania.
Ďalšie príznaky NF1
U pacientov je tendencia k podpriemernej výške a makrocefálii. Predčasná puberta je častým javom a súvisí hlavne s nádorom v oblasti chiazmy optiku. Sú popísané aj prípady oneskorenej puberty. Arteriálna hypertenzia sa vyskytuje častejšie pri NF1 než u bežnej populácie. Vaskulopatie asociované s NF1 sú dávno známe. Ide o stenózu renálnej artérie, koarktáciu aorty, aneuryzmu, chorobu Moyamoya. Rôzne formy vaskulopatie sa považujú za najčastejšiu príčinu predčasného úmrtia NF1 pacientov. Poruchy učenia, správania, pozornosti, pamäte, spánku, nižšie IQ, epilepsia následkom expanzívneho procesu CNS, autistické črty, migrenózne bolesti hlavy nie sú zriedkavými príznakmi choroby.
Diagnostické kritériá NF1
Podľa National Institutes of Health Consensus Development Center z roku 1988 ide o pacienta s neurofibromatózou 1, ak sú splnené 2 alebo viac diagnostických kritérií NF1. Každý pacient, ktorý spĺňa aspoň jedno diagnostické kritérium, je podozrivý z NF1 a mal by byť testovaný na prítomnosť mutácie v géne NF1.
Diagnostické kritériá neurofibromatózy typu 1:
6 alebo viac škvŕn typu „café-au-lait“ s rozmerom viac ako 5 mm v prepubertálnom období, respektíve nad 15 mm v postpubertálnom období,
2 alebo viac neurofibrómov akéhokoľvek typu alebo 1 plexiformný neurofibróm,
freckling v axilárnej alebo inguinálnej oblasti,
glióm optického nervu,
2 alebo viac Lischových uzlov (hamartóm dúhovky),
kostné lézie ako sfenoidálna dysplázia, non/dystrofická kyfo/skolióza, postihnutie dlhých kostí ako napríklad stenčenie kortexu, kongenitálne ohnutie, pseudoartróza predlaktia, predkolenia,
prvostupňový príbuzný, ktorý spĺňa hore uvedené diagnostické kritériá NF1.

Genetické pozadie NF1
NF1 sa dedí autozómovo dominantne. Jej výskyt nie je limitovaný na žiadnu ľudskú rasu. Frekvencia ochorenia je zhruba 1 : 3 000 – 3 500, bez uprednostnenia jedného z pohlaví. U všetkých jedincov, ktorí disponujú mutáciou pre NF1, skôr či neskôr dôjde k manifestácii ochorenia. Hovoríme teda o 100-percentnej penetrácii. NF1 „nepreskakuje“ generáciu, v postpubertálnom období neexistuje v asymptomatickej forme (nie je možné, že by ju nemal rodič, ak ju má starý rodič a vnuk). NF1 je podmienená mutáciou v géne pre neurofibromín 1. Gén sa nachádza na dlhom ramene chromozómu 17. Patrí medzi najdlhšie gény ľudského genómu. Môže sa pýšiť prvenstvom vo vzniku spontánnych mutácií počas gametogenézy. Tým sa odôvodňuje fakt, že až polovicu prípadov NF1 tvoria pacienti bez pozitívnej rodinnej anamnézy.
Samotný gén NF1 patrí medzi tumor supresorové gény. Jeho funkcia v bunkovej proliferácii zatiaľ nie je úplne prebádaná, ale jedno je isté, že aktivuje GTP-ázu, a tak tlmí RAS proteíny. Na základe týchto skutočností bola NF1 zaradená medzi RASopatie. Gén svojimi 60 exónmi patrí medzi ťažké oriešky pre molekulového genetika. Neexistujú „horúce miesta“ génu, kde sa predilekčne kumulujú mutácie. Svetové databázy evidujú viac než 1 000 rôznych mutácií, čo teda vôbec neuľahčuje vyšetrenie génu. V našom genetickom laboratóriu realizujeme sekvenačnú analýzu NF1 na úrovni genómovej DNA a detekciu delécií a duplikácií génu metódou MLPA. Záchytnosť mutácie uvedenými metódami sa pohybuje okolo 65 %. V praxi to znamená, že sa stretávame aj s rodinami, u ktorých niet pochýb o správnosti klinickej diagnózy, avšak (zatiaľ) sa nedostaneme k molekulárnemu korelátu ochorenia. Ale môžeme si byť istí, že výskum v molekulárnej genetike napreduje kozmickou rýchlosťou a čo dnes ešte nie je možné, zajtra už azda bude…
DNA analýza sa obzvlášť odporúča pacientom, ktorí majú negatívnu rodinnú anamnézu NF1 a aktuálne nespĺňajú diagnostické kritériá. Sú to predovšetkým detskí pacienti. V prípade dôkazu mutácie je však nemožné predvídať priebeh, stupeň závažnosti, prognózu a komplikácie ochorenia, keďže chýba genotypovo-fenotypová súvislosť. Záchyt mutácie v rodine je využiteľný aj pri plánovaní potomstva. Známa mutácia je detegovateľná v bunkách plodovej vody a nie je taktiež problémom vybrať to správne embryo pred transferom na základe negatívneho výsledku DNA analýzy v rámci in vitro fertilizácie.
NF2 verzus NF1
Keď sa používa termín neurofibromatóza, automaticky sa myslí na typ 1. Ďalšia, oveľa zriedkavejšia forma neurofibromatózy, je typ 2. Hovorí sa jej aj centrálna alebo bilaterálna akustická, vzhľadom na typický výskyt tumorov v CNS (bilaterálne vestibulárne schwannómy, meningeómy, ependimómy, zriedka astrocytómy), hoci uvedené nádory môžu byť prítomné aj pri NF1. Následkom tumorózneho nálezu na n. VIII začína porucha sluchu od puberty. Kožné príznaky ako škvrny café au lait, neurofibrómy a plexiformný neurofibróm sú oproti NF1 zanedbateľné, freckling, glióm n. opticus a Lischove noduly sa nevyskytujú. Pre NF2 je charateristická zadná kapsulárna katarakta. Ochorenie sa manifestuje vo veku okolo 20 rokov, hoci prvé očné a kožné prejavy sa občas hlásia už aj od detstva. Priebeh ochorenia je medzi rôznymi rodinami variabilný, v rámci jednej rodiny väčšinou podobný (pri NF1 je veľká inter aj intrafamiliárna variabilita). Benígne tumory NF2 sa vyznačujú menším potenciálom malígneho zvrhnutia, ale ich nešťastná lokalizácia môže mať fatálne následky: priemerný vek úmrtia pacienta je necelých 37 rokov!
Napriek tomu, že NF2 sa dedí aj autozómovo dominantne, NF1 a NF2 nemajú žiadny spoločný genetický základ. NF2 je podmienená mutáciou v géne NF2, ktorý sa nachádza na 22. chromozóme. Skrátka, NF2 a NF1 sú klinicky a geneticky heterogénne ochorenia.
Schwannomatóza
de o mnohopočetné schwannómy kraniálnych, miechových a periférnych nervov. Postihnutie očí, kože a schwannóm vestibulárneho nervu nie sú prítomné. Ochorenie vykazuje taktiež autozómovo dominantnú dedičnosť. Gén zodpovedný za vznik schwannomatózy je SMARCB1. Nachádza sa na rovnakom 22. chromozóme ako gén pre NF2. Okolo 85 % prípadov ochorenia vzniká na základe de novo mutácie. Pri schwannomatóze nie je zistené riziko malignizácie.
Ochorenia pripomínajúce NF1
Je to predovšetkým NF2, schwannomatóza, autozómovo dominantne dedičné mnohopočetné „café-au-lait“, Legiov syndróm, Proteov syndróm, McCuneov-Albrightov syndróm, LEOPARD syndróm, konštitučný mismatch repair deficit a iné.
Onkologické komplikácie NF1
Samotné neurofibrómy nemajú malígny potenciál. Väčšina plexiformných neurofibrómov je asymptomatická, ale niektoré môžu byť bolestivé, môžu narásť do enormných rozmerov, deformovať telo a erodovať okolité štruktúry. Plexiformný neurofibróm má nízky stupeň rizika malígnej transformácie, predovšetkým na malígny tumor obalu periférneho nervu (malignant peripheral nerve sheath tumor – MPNST). Tento typ karcinómu je najčastejšia malignita pacientov s NF1. Vyskytuje sa asi u 10 % pacientov. Vzniká predovšetkým u mladých dospelých ľudí s NF1, obzvlášť na dolných končatinách. Jeho prognóza je nepriaznivá. Gliómy optiku, mozgového kmeňa a cerebella sa správajú oveľa menej agresívne v rámci NF1 ako bez diagnózy NF1. Asi u 20 % pacientov s gliómom optickej dráhy diagnostikovaného v detstve sa počas života vyvinie ďalší glióm CNS. Bolo zistené výrazné riziko vzniku nonoptických gliómov, MPNST a choroby Moyamoya u pacientov s gliómom v súvislosti s predošlou rádioterapiou. Nie je zanedbateľné ani vyššie riziko vzniku ďalších tumorov ako u bežnej populácie. Ide napríklad o karcinóm prsníka, feochromocytóm, Wilmsov tumor, juvenilnú chronickú myeloidnú leukémiu, myelodysplastické syndrómy, karcinoid duodena, gastrointestinálne stromálne tumory, retinálne vasoproliferatívne tumory, nediferencované sarkómy a neuroblastómy.
Dispenzarizácia pacienta s NF1
Multiorgánové postihnutie pri NF1 vyžaduje úzku spoluprácu viacerých špecialistov (dermatológ, neurológ, oftalmológ, otorinolaringológ, psychológ, ortopéd, radiológ, kardiológ, atď.) U detí sa starostlivo zameriavame na psychomotorický vývoj, školský prospech, nástup puberty, výšku, hmotnosť, obvod hlavy a tlak krvi. Ženám, ktoré užívajú antiepileptiká v prekoncepčnom období a na začiatku gravidity, sa odporúča užívanie Acidum folicum v dennej dávke 5 mg (táto dávka je mnohokrát väčšia než dávka odporúčaná zdravým ženám – t. j. 0,4 mg). Veľký neurofibróm v pelvickej oblasti môže byť prekážkou prirodzeného pôrodu.
Terapia NF1
Neurofibrómy sa vo všeobecnosti neexcidujú, obzvlášť neurofibrómy malých rozmerov. Ak sú na nevhodnom mieste alebo extrémnych rozmerov, odstraňujú sa chirurgicky alebo laserom. Chirurgické riešenie plexiformných neurofibrómov je neuspokojivé. Často dochádza k recidívam. V súčasnosti prebieha viacero klinických štúdií liečby plexiformných neurofibrómov, napríklad imatinibom, pegylovaným interferónom-alfa-2b, s nádejnými výsledkami. Progredujúce gliómy n. opticus sa riešia chemoterapeutikami (carboplatin, vincristin) a chirurgicky. Rádioterapia gliómov a plexiformných neurofibrómov je kontraindikovaná pre už vyššie spomínané riziko vzniku choroby moyamoya, MPNST a následných gliómov. Riešením operabilného MPNST je chirurgické odstránenie, niekedy až s potrebou amputácie končatiny vzhľadom na časté recidívy. Chemoterapia neresekabilného a metastatického MPNST ostáva kontroverzná kvôli nedostatočnému efektu. Preto sa testujú farmaká zasahujúce do dráhy RAS ako farnesyl- transferáza v kombinácii s lovastatínom, sorafenib, rapamycín-komplex-1-inhibítor spolu s erlotinibom či MEK inhibítory.
Záver
Zatiaľ neexistuje štandardizovaná terapia NF1 a jej komplikácií. Prebiehajú štúdie, ktoré majú za cieľ objektivizovať možný (ne)úspech liečby na základe genotypu pacienta, a tým vytvoriť terapiu šitú na mieru. NF1 pre jej komplexnosť ostáva na platforme terapie naďalej výzvou.
Literatúra
1. www.genetests.org
2. National Institute of Health Consensus Development Conference. Neurofibromatosis: Conference Statement. Arch Neurol Chicago 1988; 45: 575 – 578
3. Petrák B., Plevová P., Novotný J., Foretová L. Neurofibromatosis von Recklinghausen. Klin Onkol 2009; 22(Suppl): S38 – S44




