Šíriaci sa koronavírus SARS 2 (SARS-CoV-2) predstavuje obrovskú hrozbu pre ľudské zdravie. Zhang, Wu a i. ukázali, že podobne ako netopiere, aj druhy šupinavca sú prírodným zdrojovým druhom koronavírusov podobných SARS-CoV-2. Toto zistenie by mohlo pomôcť nájsť prechodného hostiteľa SARS-CoV-2, aby sa mohla celosvetová koronavírusová pandémia zastaviť.
Úvodné zhrnutie
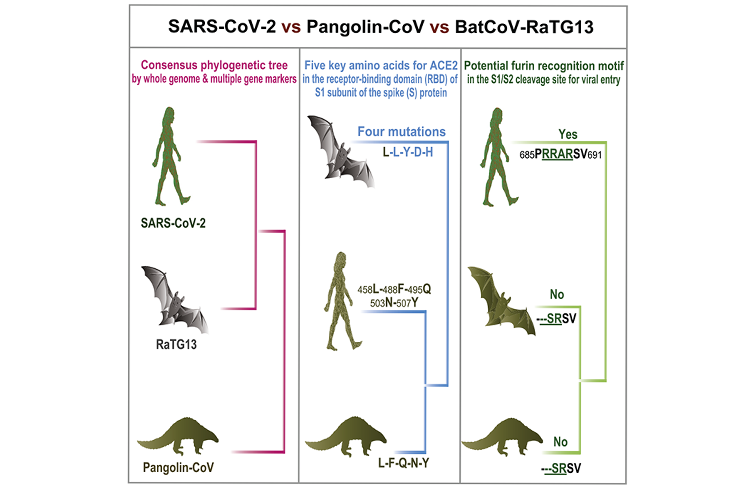
Prepuknutie koronavírusovej choroby 2019 (COVID-19) spôsobenej novým koronavírusom 2019 (SARS-CoV-2) začalo v meste Wuhan v Číne a rozšírilo sa do celého sveta. V súčasnosti je životne dôležité nájsť potenciálnych prechodných hostiteľov SARS-CoV-2, aby sa šírenie choroby COVID-19 dostalo pod kontrolu. Z týchto dôvodov sme opäť preskúmali publikované údaje o vzorkách pľúc šupinavca, v ktorých koronavírusy podobné SARS-CoV našli Liu a kol. [1]. U mŕtvych šupinavcov v Malajzii sme našli genómové a vývojové dôkazy o výskyte koronavírusu podobného SARS-CoV-2 (nazvaného šupinavčí CoV). Vírus CoV zo šupinavca je z 91,02 % identický so SARS-CoV-2 na celej úrovni genómu a z 90,55 % identický s BatCoV RaTG13 z netopiera. Popri RaTG13 je CoV zo šupinavca najbližším príbuzným CoV k vírusu SARS-CoV-2. Proteín S1 z CoV zo šupinavca je omnoho viac príbuzný k vírusu SARS-CoV-2 než k RaTG13. Zvyšky piatich kľúčových aminokyselín zapojených v interakcii s ľudskými proteínmi ACE2 sú medzi vírusom CoV zo šupinavca a SARS-CoV-2 úplne konzistentné, no v RaTG13 sú prítomné štyri mutácie aminokyselín. Oba vírusy, CoV zo šupinavca aj RaTG13, stratili zdanlivý motív rozpoznávacej sekvencie furínov v mieste štiepenia S1/S2, čo sa dá pozorovať v SARS-CoV-2. Táto štúdia nezvratne tvrdí, že druhy šupinavca sú prírodným zdrojovým druhom koronavírusov podobných SARS-CoV-2.
Výsledky a diskusia
Podobne ako v prípade SARS-CoV a MERS-CoV [2], netopier je stále pravdepodobným druhom pôvodu nového koronavírusu 2019 (SARS-CoV-2), lebo SARS-CoV-2 má z 96 % celogenómovú identitu s koronavírusom z netopiera BatCoV RaTG13, z netopiera Rhinolophus affinis z provincie Yunnan [3]. SARS-CoV a MERS-CoV však obvykle prechádzajú na prechodných hostiteľov, ako sú cibetky alebo ťavy a až potom prechádzajú na ľudí [4]. Táto skutočnosť naznačuje, že SARS-CoV-2 bol na ľudí pravdepodobne prenesený inými živočíchmi. Ak uvážime, že pacient pri prvej koronavírusovej chorobe 2019 (COVID-19) nehlásil žiadnu expozíciu na trhu s morskými plodmi [5], je životne dôležité nájsť prechodného hostiteľa SARS-CoV-2, aby sa zastavil prenos medzi druhmi. Dňa 24. októbra 2019 Liu a jeho kolegovia z čínskeho Strediska záchrany divých zvierat v provincii Guangdong [1] ako prví odhalili existenciu koronavírusu podobného SARS-CoV vo vzorkách pľúc dvoch mŕtvych malajských šupinavcov so spenenou tekutinou v pľúcach a pľúcnou fibrózou a tento fakt objavili zhruba okolo času prepuknutia COVID-19. Použitím ich publikovaných výsledkov sme ukázali, že všetky vírusové kontigy (súbor prekrývajúcich sa klonov z genómovej knižnice) zostavené z dvoch pľúcnych vzoriek (lung07 a lung08) vykazovali nízke identity, počnúc od 80,24 % po 88,93 %, so známymi koronavírusmi SARS-CoV. Domnievame sa teda, že mŕtve malajské šupinavce môžu prenášať nový CoV, ktorý je blízky príbuzný so SARS-CoV-2.
Hodnotenie pravdepodobnosti prítomnosti CoV podobného SARS-CoV-2 v rode šupinavcov
S cieľom potvrdiť náš predpoklad sme načítali dáta o sekvenovaní nespracovanej RNA (RNA-seq) (SRA: PRJNA573298) pre tieto dve pľúcne vzorky z archívu sledov sekvencií SRA a vykonali sme kontrolu kvality a odstránenie kontaminantov zhodné s tým, ako opísal v štúdii Liu [1]. Našli sme 1 882 čistých sledov zo vzorky lung08, ktorá bola nanesená na referenčný genóm SARS-CoV-2 (GenBank: MN908947) [6] a pokrývala 76,02 % genómu SARS-CoV-2. Vykonali sme zostavenie týchto sledov de novo a získali sme 36 kontigov s dĺžkami od 287 bp po 2 187 bp, s priemernou dĺžkou 700 bp. Analýzou s databázami Blast v porovnaní s bielkovinám z 2 845 referenčných genómov CoV vrátane RaTG13, SARS-CoV-2s a iných známych CoV sme zistili, že 22 kontigov najlepšie zodpovedalo koronavírusom SARS-CoV-2 (identita aminokyselín zo 70,6 % až 100 %; priemer: 95,41 %) a že 12 kontigov zodpovedalo koronavírusu podobnému SARS-CoV netopiera (identita aminokyselín z 92,7 % až 100 %; priemer: 97,48 %) (tabuľka S1). Tieto výsledky naznačujú, že malajský šupinavec by mohol prenášať nový CoV (tu nazvaný šupinavčí CoV), ktorý je podobný SARS-CoV-2.
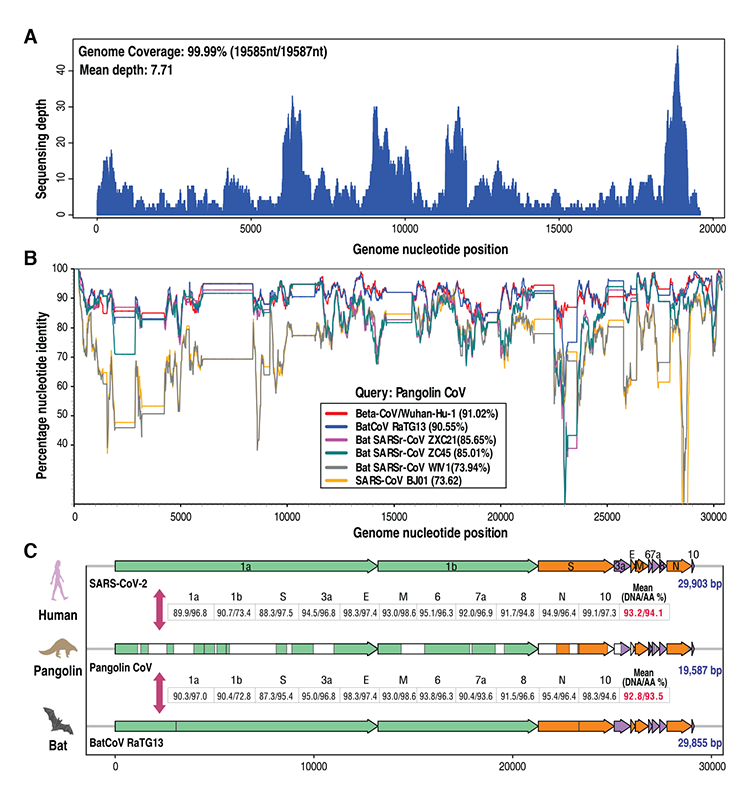
Návrh genómu šupinavčieho CoV a jeho genómové charakteristiky
Použitím podporného prístupu riadeného odkazmi sme na základe vyššie uvedených 34 kontigov vytvorili návrh genómu CoV šupinavca (19 587 bp). S cieľom znížiť vplyv chýb nespracovaných sledov na kvalitu podpory boli ručne vyradené malé fragmenty zoradené proti referenčnému genómu s dĺžkou menej než 25 bp, ak sa nedali pokryť veľkými fragmentmi alebo referenčným genómom. Opätovné nanášanie 1 882 sledov v porovnaní s návrhom genómu prinieslo pokrytie genómov 99,99 % (rozsah pokrytia: 1X–47X) (obrázok č. 1 – A). Priemerné pokrytie predstavovalo v rámci celého genómu 7,71X, čo je dvojnásobne vyššie než najnižšie spoločné pokrytie sledov 3X pre vyvolanie SNP na základe sekvenovania nízkeho pokrytia v pilotnej fáze projektu 1 000 genómov [7]. Podobné úrovne pokrytia sú tiež dostačujúce na odhalenie zriedkavých mikrobiálnych druhov alebo mikrobiálnych druhov s nízkou početnosťou z metagenómových databáz [8], čo naznačuje, že náš návrh genómu CoV šupinavca sa dá spoľahlivo ďalej analyzovať. Pri analýze Simplot [9] vykazoval v rámci celého genómu CoV šupinavca vysokú celkovú identitu sekvencií genómu s RaTG13 (90,55 %) a SARS-CoV-2 (91,02 %) (Obrázok č. 1 – B), hoci vyššia identita (96,2 %) bola medzi SARS-CoV-2 a RaTG13 [3]. Inými koronavírusmi podobnými SARS-CoV, podobné šupinavčiemu CoV, boli netopierí SARSr-CoV ZXC21 (85,65 %) a netopierí SARSr- CoV ZC45 (85,01 %). Kým sa táto práca hodnotila, dve podobné štúdie v predtlači zistili, že koronavírusy u šupinavcov zdieľali identitu DNA na 90,3 % [10] a 92,4 % [11] so SARS-CoV-2, čo znamená približne 91,02-percentnú identitu so SARS-CoV-2 pozorovanú v tomto dokumente a podporujúcu naše zistenia. Ak tieto výsledky zlúčime, naznačuje to, že šupinavčí CoV by mohol mať spoločný pôvod so SARS-CoV-2 a RaTG13. Organizácia genómu šupinavčieho CoV bola charakterizovaná zarovnaním sekvencií v porovnaní so SARS-CoV-2 (GenBank: MN908947) a RaTG13. Genóm CoV šupinavca obsahuje šesť väčších otvorených čítacích rámcov (ORF) spoločných pre koronavírusy a štyri ďalšie doplnkové gény (Obrázok č. 1 − C; Obrázok č. 2). Ďalšia analýza naznačila, že gény CoV šupinavca boli zarovnané s génmi SARS-CoV-2 s pokrytím od 45,8 do 100 % (priemerné pokrytie 76,9 %). Gény CoV šupinavca zdieľali vysokú priemernú identitu nukleotidov a aminokyselín s oboma druhmi génov: SARS-CoV-2 (GenBank: MN908947) (identita nukleotidov 93,2 %/identita aminokyselín 94,1 %) aj RaTG13 (identita nukleotidov 92,8 % / identita aminokyselín 93,5 %) (Obrázok č. 1 − C; Obrázok č. 2). Je prekvapujúce, že niektoré gény CoV šupinavca vykazovali vyššiu identitu sekvencií aminokyselín s génmi SARS CoV-2 než s génmi RaTG13, vrátane orf1b (73,4 %/72,8 %), proteínu S-(„spike“) (97,5 %/95,4 %), orf7a (96,9 %/93,6 %) a orf10 (97,3 %/94,6 %). Vyššia identita aminokyselín proteínu S udeľuje funkčnú podobnosť medzi CoV šupinavca a SARS CoV-2.
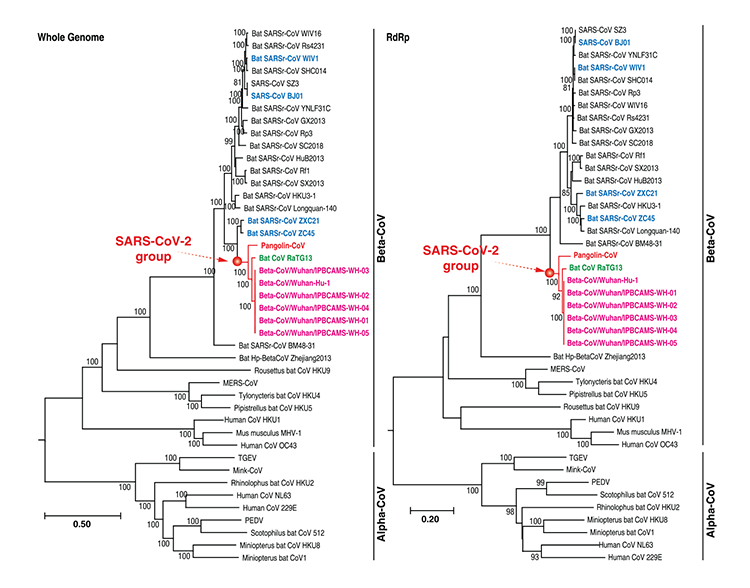
Fylogenetické vzťahy koronavírusov šupinavca, RaTG13 a SARSCoV-2
S cieľom stanovenia evolučných vzťahov medzi CoV šupinavca, SARS-CoV-2, a predtým identifikovaných koronavírusov sme pripravili fylogenetické stromy na báze sekvencií nukleotidov celogenómovej sekvencie, génu polymerázy RNA-dependentnej RNA (RdRp), neštrukturálnych proteínových génov ORF1a a ORF1b a hlavných štrukturálnych proteínov kódovaných génmi S a M. Vo všetkých fylogenetických stromoch sme združili CoV šupinavca, RaTG13, a SARS-CoV-2 do dobre podporovanej skupiny, nazvanej v tomto dokumente „skupina SARS-CoV-2“ (Obrázok č. 2). Táto skupina predstavuje skupinu nových betakoronavírusov. V rámci tejto skupiny boli zoskupené dohromady RaTG13 a SARSCoV- 2 a CoV šupinavca bol ich najpríbuznejším spoločným predkom. Otázku, či pre SARS-CoV-2 je bazálnou skupinou SARSr-CoV ZXC21 a/alebo SARSr- CoV ZC45, však ešte stále treba prediskutovať. Taká diskusia sa vyskytla aj v oboch štúdiách: autorov Wu a kol. [6] aj Zhou a kol. [3]. Možným vysvetlením je dávny prípad rekombinácie v skupine betakoronavírusov [6]. Treba zmieniť, že odhalené evolučné vzťahy koronavírusov ukázané celým genómom, génom RdRp a génom S sú vysoko konzistentné so vzťahmi vykazovanými úplnými genómovými informáciami v štúdii autorov Zhou a i. [3]. Táto zhoda naznačuje, že náš návrh genómu CoV šupinavca obsahuje dostatočnú genómovú informáciu, aby bolo možné sledovať skutočnú vývojovú pozíciu CoV šupinavca medzi koronavírusmi.
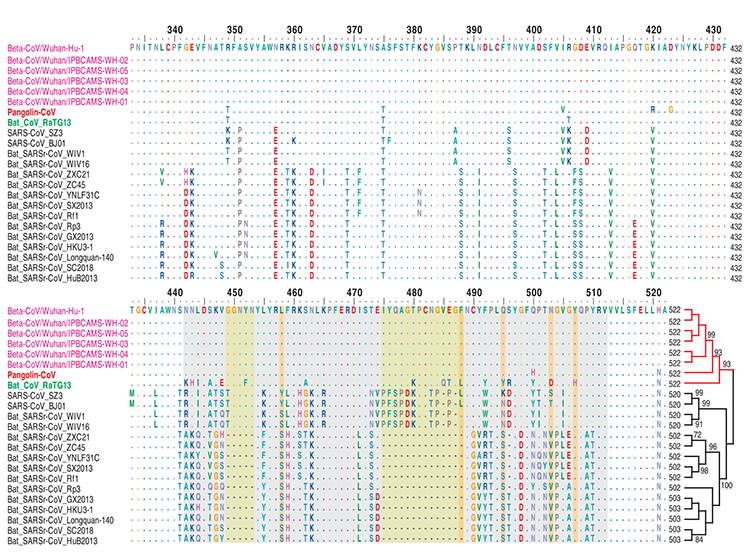
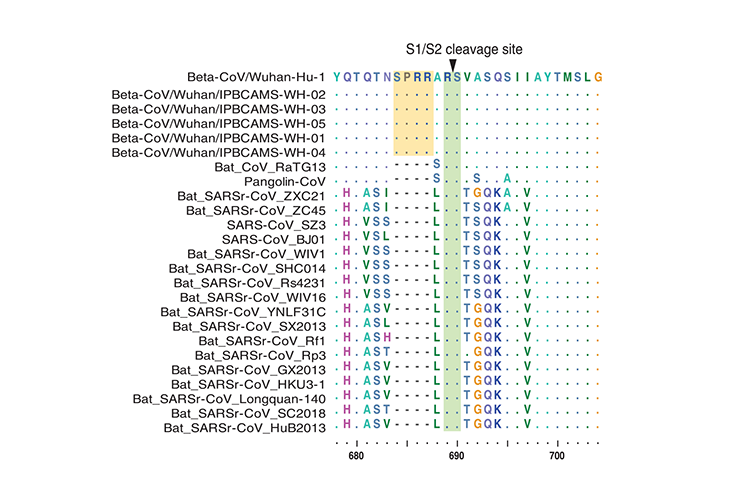
Dualizmus proteínu S koronavírusu šupinavca
Proteín S CoV sa skladá z dvoch podjednotiek (S1 a S2), sprostredkúva infekciu hostiteľských buniek exprimujúcich receptory a je kľúčový. S1 obsahuje doménu viažucu sa na receptory (RBD) obsahujúcu fragment asi zo 193 aminokyselín, ktorý zodpovedá za rozpoznávanie receptora na povrchu buniek a väzbu [13, 14]. Zhou a kol. experimentálne potvrdili, že SARS-CoV-2 dokáže používať bielkoviny ACE2 človeka, netopiera vrápenca, cibetky a ošípanej ako vstupný receptor v bunkách exprimujúcich ACE2 [3], naznačujúc, že RBD SARS-CoV-2 sprostredkuje infekciu u ľudí a ďalších živočíchov. S cieľom získať náhľad na patogénny potenciál CoV šupinavca až na úrovni sekvencií sme prvý raz skúmali model variácií aminokyselín proteínov S1 z CoV šupinavca, SARS-CoV-2, RaTG13 iných reprezentatívnych koronavírusov SARS/SARSr. Aminokyselinový fylogenetický strom ukázal, že proteín S1 CoV šupinavca je viac príbuzný takémuto stromu CoV 2019 než stromu RaTG13. V rámci domény viažucej sa na receptor RBD sme ďalej našli, že CoV šupinavca a SARS-CoV-2 boli vysoko zachované, len s jedinou aminokyselinovou zmenou (500H/500Q) (Obrázok č. 3),ktorá nie je jedným z piatich kľúčových zvyškov zapojených do interakcie s ľudským ACE2 [3, 14]. Tieto výsledky naznačujú, že CoV šupinavca by mohol mať patogénny potenciál podobný ako pri SARS-CoV-2. Na rozdiel od toho, RaTG13 má zmeny v 17 aminokyselinových zvyškoch, z ktorých sú 4 medzi kľúčovými aminokyselinovými zvyškami (Obrázok č. 3). Existujú dôkazy naznačujúce, že zmena 472L (SARS-CoV) na 486F (SARS-CoV-2) (zodpovedajúca zmene druhého kľúčového aminokyselinového zvyšku na Obrázku č. 3) môže posilniť van der Waalsovo spojenie s M82 (ACE2) [15]. Popri tom, dôležitá substitúcia 404V v RBD SARS-CoV použitím 417K v RBD SARSCoV- 2 (pozri polohu zarovnania 420 na Obrázku č. 3 a bez zmeny aminokyseliny medzi SARS-CoV-2 a RaTG13) môže viesť k tesnejšiemu spojeniu z dôvodu vzniku soľných mostíkov medzi 417K a 30D proteínu ACE2 [15]. Aj tak je však ešte stále potrebný ďalší výskum toho, či tieto mutácie majú vplyv na afinitu pre ACE2. To, či sú CoV šupinavca alebo RaTG13 potenciálnymi infekčnými agensami pre človeka, treba ešte zistiť. Miesto štiepenia S1/S2 v proteíne S je tiež dôležitým určujúcim prvkom prenosnosti a patogenity vírusov SARSCoV/ SARS-CoVr [16] Počas infekcie sa trimetrický proteín S transformuje proteázami hostiteľských buniek v mieste štiepenia S1/S2. Po štiepení alebo tzv. stimulácii sa proteín delí na N-terminálnu S1-ektodoménu, ktorá rozpoznáva príbuzný receptor na povrchu buniek a C-terminálny proteín ukotvený na S2-membránu, ktorý riadi zlúčenie obalu vírusu s bunkovou membránou. Zistili sme, že proteín S vírusu SARS-CoV-2 obsahuje zdanlivý motív rozpoznávacej sekvencie furínov (PRRARSV) (obrázok 4) podobný motívu v MERS-CoV, ktorý má motív PRSVRSV pravdepodobne štiepený furínom [16, 17] počas uvoľnenia vírusov. Na druhej strane, furínový sekvenčný motív v mieste S1/S2 chýba v proteíne S CoV šupinavca a vo všetkých iných koronavírusoch SARS/SARSr. Tento rozdiel naznačuje, že SARS-CoV-2 mohol získať iný mechanizmus na podporu vstupu do hostiteľských buniek [18]. Je zaujímavé, že okrem koronavírusu MERS-CoV boli podobné vzory sekvencií ako v SARS-CoV-2 prítomné aj u niektorých členov podskupín alfakoronavírusov, betakoronavírusov a gamakoronavírusov [19], čo vedie k zaujímavej otázke, či by sa tento furínový sekvenčný motív v SARSCoV-2 dal odvodiť od existujúcich proteínov S iných koronavírusov alebo prípadne, či by SARS-CoV-2 mohol byť rekombinantom z CoV šupinavca alebo RaTG13 a ďalších koronavírusov s podobným motívom rozpoznávania furínov u neznámeho prechodného hostiteľa.
Variácie aminokyselín v nukleokapsidovom (N) proteíne na prípadné diagnostikovanie
Proteín N je najhojnejším proteínom v koronavírusoch. Proteín N je vysoko imunogénnym fosfoproteínom a bežne je veľmi zachovaný. Proteín N CoV sa často používa ako marker na diagnostické testy. S cieľom získať ďalší náhľad na diagnostický potenciál CoV šupinavca sme skúmali model variácií aminokyselín proteínov N z CoV šupinavca, SARS-CoV-2, RaTG13 iných reprezentatívnych koronavírusov SARS/SARSr. Fylogenetická analýza na báze proteínu N podporila klasifikáciu CoV šupinavca ako sesterského taxónu SARS-CoV-2 a RaTG13 (Obrázok č. 3). Ďalej sme našli sedem aminokyselinových mutácií, ktoré odlišovali naše definované koronavírusy „skupiny SAR-CoV-2“ (12N, 26 G, 27S, 104D, 218A, 335T, 346N a 350Q) od iných známych SARS-CoV (12S, 26D, 27N, 104E, 218T, 335H, 346Q a 350N). Dve aminokyselinové miesta (38P a 268Q) sú zdieľané CoV šupinavca, RaTG13 a SARS-CoV, ktoré sú mutované na 38S a 268A v SARS-CoV-2. Len jediný aminokyselinový zvyšok zdieľaný CoV šupinavca a SARS-CoV (129E) sa zhodne líši v SARS-CoV-2 aj RaTG13 (129D). Pozorované zmeny aminokyselín v proteíne N by mohli byť užitočnými pri vyvíjaní antigénov s vyššou citlivosťou pre sérologickú detekciu SARS-CoV-2.
Záver
Na základe publikovaných metagonómových dát táto štúdia poskytuje prvú správu o potenciálnom blízkom príbuzenstve (CoV šupinavca) SARS-CoV-2, ktorý bol objavený v mŕtvych malajských šupinavcoch po značnom úsilí o záchranu. Popri RaTG13 je CoV zo šupinavca najbližším príbuzným CoV k vírusu SARS-CoV-2. Vzhľadom na nedostupnosť pôvodnej vzorky sme nevykonali ďalšie pokusy, aby sme svoje zistenia mohli potvrdiť (validačnou PCR, sérologickou detekciou alebo dokonca izoláciou častíc vírusu). Genóm CoV šupinavca, ktorý sme objavili, vykazoval identitu nukleotidov s genómom SARS-CoV-2 rovnú 91,02 %. Avšak to, či sú druhy šupinavcov dobrými kandidátmi na pôvodcu SARS-CoV-2, treba ešte prediskutovať. Ak zvážime veľké rozšírenie SARS-CoV v prírodných zdrojových druhoch, ako sú netopiere, ťavy a šupinavce, naše zistenia by mohli byť cenným prínosom pre nájdenie nových prechodných hostiteľov SARS-CoV-2, aby sa zastavil prenos medzi druhmi.
Pozn. red.: Publikované v Current Biology, apríl 2020. Preložené pre inVitro s láskavým súhlasom autorov v snahe oboznámiť čo najširšiu odbornú verejnosť s ich poznatkami v čase celosvetovej pandémie nového koronavírusu.
Poďakovanie Túto štúdiu podporila Second Tibetan Plateau Scientific Expedition and Research (STEP) (č. 2019QZKK0503), National Key Research and Development Program of China (č. 2018YFC2000500), Key Research Program of the Chinese Academy of Sciences (č. KFZDSW-219) a Chinese National Natural Science Foundation (č. 31970571).
Autorské príspevky Z.Z. vykonal plánovanie, koordináciu, realizáciu a facilitáciu projektu. T.Z. a Q.W. vykonali metagenomickú analýzu. T.Z. zrealizovala zostavy, génovú predpoveď a anotáciu. Q.W. spracoval zozbierané údaje a fylogenetickú analýzu. Z.Z., T.Z. a Q.W. pripravil rukopis.
Literatúra
Liu, P., Chen, W., and Chen, J.-P. (2019). Viral metagenomics revealed sendai virus and coronavirus infection of Malayan Pangolins (Manis javanica). Viruses 11, 979.
Li, W., Shi, Z., Yu, M., Ren, W., Smith, C., Epstein, J.H., Wang, H., Crameri, G., Hu, Z., Zhang, H., et al. (2005). Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679.
Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., Si, H.-R., Zhu, Y., Li, B., Huang, C.-L., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. Published online February 3, 2020. https://doi.org/10.1038/s41586-020-2012-7.
Cui, J., Li, F., and Shi, Z.-L. (2019). Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192.
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506.
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature. Published online February 3, 2020. https://doi.org/10.1038/s41586-020-2008-3.
Abecasis, G.R., Altshuler, D., Auton, A., Brooks, L.D., Durbin, R.M., Gibbs, R.A., Hurles, M.E., and McVean, G.A.; 1000 Genomes Project Consortium (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073.
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K.L., Tyson, G.W., and Nielsen, P.H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538.
Lole, K.S., Bollinger, R.C., Paranjape, R.S., Gadkari, D., Kulkarni, S.S., Novak, N.G., Ingersoll, R., Sheppard, H.W., and Ray, S.C. (1999). Fulllength human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160.
Xiao, K., Zhai, J., Feng, Y., Zhou, N., Zhang, X., Zou, J.-J., Li, N., Guo, Y., Li, X., Shen, X., et al. (2020). Isolation and characterization of 2019-nCoVlike coronavirus from Malayan pangolins. bioRxiv. https://doi.org/10.1101/2020.02.17.951335.
Lam, T.T.-Y., Shum, M.H.-H., Zhu, H.-C., Tong, Y.-G., Ni, X.-B., Liao, Y.-S., Wei, W., Cheung, W.Y.-M., Li, W.-J., Li, L.-F., et al. (2020). Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. bioRxiv. https://doi.org/10.1101/2020.02.13.945485.
Tortorici, M.A., and Veesler, D. (2019). Structural insights into coronavirus entry. In Advances in Virus Research, F.A. Rey, ed. (Academic Press), pp. 93–116.
Ge, X.-Y., Li, J.-L., Yang, X.-L., Chmura, A.A., Zhu, G., Epstein, J.H., Mazet, J.K., Hu, B., Zhang, W., Peng, C., et al. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538.
Wong, S.K., Li, W., Moore, M.J., Choe, H., and Farzan, M. (2004). A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279, 3197–3201.
Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y., and Zhou, Q. (2020). Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. Published online March 4, 2020. https://doi.org/10.1126/science.abb2762. 16.
Millet, J.K., and Whittaker, G.R. (2014). Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 111, 15214–15219.
Coutard, B., Valle, C., de Lamballerie, X., Canard, B., Seidah, N.G., and Decroly, E. (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 176, 104742.
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kru¨ ger, N., Herrler, T., Erichsen, S., Schiergens, T.S., Herrler, G., Wu, N.-H., Nitsche, A., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. Published online March 4, 2020. https://doi.org/10.1016/j.cell.2020.02.052.
Millet, J.K., and Whittaker, G.R. (2015). Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 202, 120–134.
Bolger, A.M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120.
Langmead, B., and Salzberg, S.L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359.
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, T.L. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., and Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079.
Edgar, R.C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797.
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549.
Tento článok sa nachádza v čísle invitro 02/2020
Koronavírus
Obsah druhého a zároveň špeciálneho čísla roka 2020 je venovaný novému koronavírusu SARS-CoV-2. Aj v tomto čísle však nájdete naše tradičné rubriky – rozhovory so zaujímavými ľuďmi, články…