

Endokrinologické ochorenia sa prejavujú zníženou alebo zvýšenou tvorbou hormónov. Každý hormón v tele má špecifickú funkciu a jeho hladina je nastavená a udržiavaná tak, ako je v danej chvíli potrebné pre optimálne fungovanie organizmu.
Didaktické členenie na úvod
Nádory v endokrinológii možno rozdeliť na 2 skupiny:
- nádory nachádzajúce sa v samotných endokrinných žľazách – produkujúce hormóny alebo hormonálne inaktívne
- a nádory produkujúce hormóny mimo endokrinných žliaz.
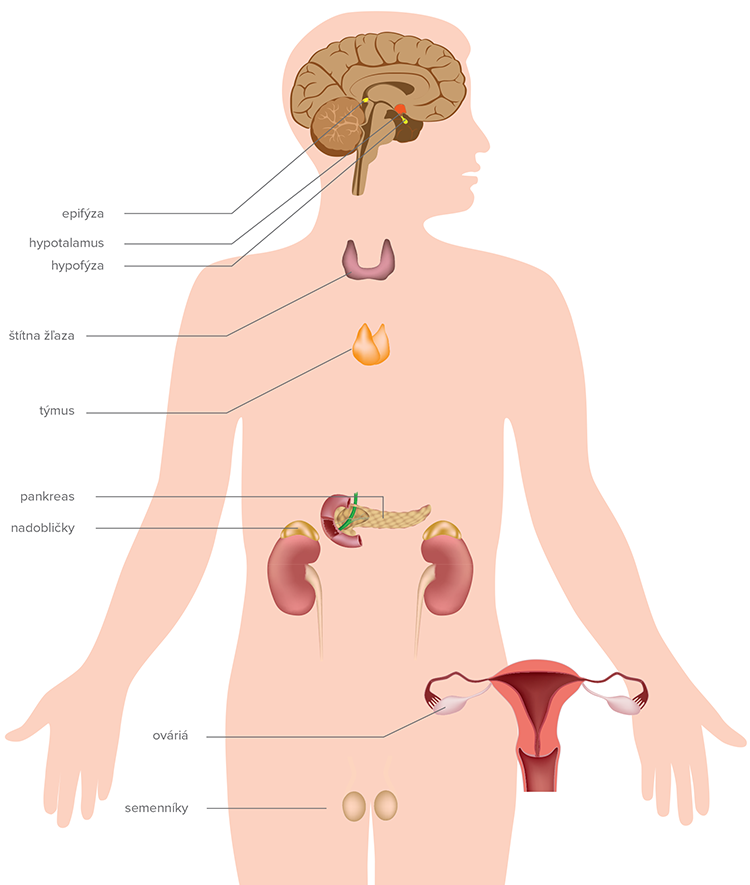
Žľazy s vnútornou sekréciou endokrinnej štruktúry sú:
- hypofýza,
- štítna žľaza,
- nadobličky,
- gonády (testes, ováriá),
- prištítne telieska,
- pankreas,
- pečeň,
- obličky,
- srdce,
- epifýza,
- GIT,
- cievny endotel,
- difúzny endokrinný systém,
- placenta.
Hypofýza
K fyziologickej hyperplázii adenohypofýzy dochádza v puberte, tehotenstve a pri laktácii. Nádory hypofýzy – tvoria 12 % nádorov CNS a sú lokalizované prevažne v adenohypofýze, menej zriedkavo v neurohypofýze (produkcia ADH, oxytocínu). Nádory hypofýzy tvoria väčšinou adenómy. Karcinómy hypofýzy sú vzácne – väčšinou afunkčné alebo deštrukčné s hypofunkciou.
Hyperpituitarizmus je súborné označenie pre nadmernú sekréciu hormónov adenohypofýzy. Hyperpituitarizmus je vždy parciálny a ide o nadprodukciu 1 – 2 hormónov tvoriacich sa v hypofýze – adenohypofýze. U väčšiny pacientov je príčinou hyperpituitarizmu adenóm hypofýzy.
Pravdepodobne jestvuje aj primárna hypotalamová forma hyperpituitarizmu, pri ktorej nadmerná stimulácia pochádzajúca z hypotalamu vedie najprv k hyperplázii trofných buniek hypofýzy a neskôr k adenomatóznej prestavbe hyperplastického tkaniva.
Hypersekrécia prolaktínu je najčastejšou formou hyperpituitarizmu a jednou z príčin hypogonadizmu. Patologická hyperprolaktinémia je opakovane zachytená zvýšená hodnota prolaktínu, po vylúčení gravidity a laktácie (fyziologické zvýšenie nastáva aj pri strese, nadmernej fyzickej aktivite, cvičení, deprivácii spánku, koite).
Patologické príčiny hyperprolaktinémie sú:
- ochorenia hypotalamu – nádory, infiltratívne ochorenia, pseudotumory, ožiarenie, trauma hlavy, MTS,
- ochorenia hypofýzy – prolaktinómy:
– nádory so zmiešanou sekréciou PRL a GH (akromegália) alebo PRL a ACTH (Cushingova choroba),
– prerušenie hypofyzárnej stopky (mechanické, funkčné),
– syndróm prázdneho tureckého sedla,
– iné nádory s útlakom na stopku hypofýzy
(nesekretorické adenómy hypofýzy, meningeómy, MTS, germinóm, kraniofaryngeóm),
– infiltratívne ochorenia (sarkoidóza, obrovskobunkový granulóm, autoimúnna lymfocytárna hypofyzitída), - lieky – psychofarmaká – antipsychotiká,
- endokrinné ochorenia – primárna hypotyreóza, insuficiencia kôry nadobličiek, Sheehanov syndróm,
- metabolické ochorenia – chronická renálna insuficiencia, hepatopatia,
- neurogénne príčiny – dráždenie hrudnej steny, genitálnej oblasti, lézie miechy,
- epilepsia,
- mentálna anorexia,
- stres – infarkt myokardu, operácia, hypoglykémia,
- zhubné nádory – ektopická tvorba prolaktínu v nádoroch, zvýšená tvorba prolaktínu v hypofýze sprevádzajúca nádory,
- gynekologické príčiny – syndróm polycystických ovárií (PCOS), myóm maternice, endometrióza,
- autoimunitné ochorenia – systémový lupus, reumatoidná artritída, sclerosis multiplex, psoriatická artritída, autoimunitná tyreoiditída, lymfocytárna hypofyzitída, autoimúnna insuficiencia kôry nadobličiek, transplantačná rejekcia, diabetes mellitus 1. typu,
- autoprotilátky proti prolaktínu – makroprolaktinémia,
- idiopatická hyperprolaktinémia – intermitentná alebo latentná
V diagnostike hypeprolaktinémie majú nezastúpiteľné miesto anamnéza, fyzikálne vyšetrenie, laboratórne stanovenie bazálneho PRL, prípadne makroprolaktínu, vyšetrenie fT4, TSH, gynekologických markerov, kortizolu, renálnych a hepatálnych testov.
Rádiologická diagnostika – MR hypofýzy a mozgu s aplikáciou gadolínia je preferované pred CT s i. v. kontrastom. Zároveň je potrebné vyšetrenie perimetra a očného pozadia oftalmológom.
Hypofyzárne adenómy zložené z laktotropných buniek hypofýzy, ktoré produkujú prolaktín, tvoria 40 % pituitárnych adenómov. U dospelých je prevalencia 100/1 milión, u detí 0,1/1 milión. Pomer žien k mužom je 10 : 1.
Podľa veľkosti delíme nádory produkujúce prolaktín na:
- mikroprolaktinómy – veľkosť do 10 mm,
- makroprolaktinómy – nad 10 mm,
- obrovské prolaktinómy nad 40 mm a/alebo väčšie ako 20 mm so supraselárnou expanziou.
Podľa správania tieto nádory delíme na benígne a malígne, malígne nádory sú veľmi zriedkavé. Ich príčinami sú hypotalamická dysfunkcia a včasná génová mutácia s následnou mutáciou a proliferáciou kmeňovej bunky hypofýzy.
Typická klinická symptomatológia pre hyperprolaktinémiu a prolaktinóm u žien sa prejavuje oligomenoreou, amenoreou, poruchou luteálnej fázy menštruačného cyklu, anovuláciou, galaktoreou, hirzutizmom, osteoporózou, inzulínovou rezistenciou, prírastkom na hmotnosti, hyperkoagulačným stavom a depresiou. U mužov je typická porucha libida, potencie, infertilita, oligo/azoospermia, hypogonadizmus, gynekomastia, galaktorea a rovnako ako u žien osteoporóza a hyperkoagulačný stav. U detí nachádzame oneskorenú pubertu, primárnu amenoreu u dievčat, poruchu rastu a galaktoreu.
Komplikáciami pri týchto – ako aj ostatných tumoroch hypofýzy – sú útlakový syndróm s cefaleou, cerebrálnymi a psychickými poruchami, poruchy perimetra, parézy hlavových nervov, poruchy hypofyzárnej funkcie, likvorea a hydrocefalus.
Liečba prolaktinómov
1. Medikamentózna liečba
Liekom voľby sú agonisti dopamínu (bromokryptín, kabergolín, quinagolid), ktoré stimuláciou D2 receptorov laktotropných buniek hypofýzy znižujú tvorbu a sekréciu prolaktínu a vedú k redukcii objemu prolaktinómu.
2. Chirurgická liečba
Až po zlyhaní medikamentóznej liečby (nedostatočné zníženie PRL bez úpravy gonadálnej funkcie, zväčšenie tumoru, rast tumoru napriek dostatočnej redukcii PRL). Ak sa pacientka snaží otehotnieť a predchádzajúca gravidita bola komplikovaná symptomatickou expanziou tumoru. Symptomatické nestabilné zväčšenie tumoru počas gravidity neodpovedajúce na opätovné zahájenie liečby agonistami dopamínu.
Chirurgické odstránenie prolaktinómu sa uskutočňuje selektívnou transsfenoidálnou adenotómiou. Väčšina mikroadenómov a makroadenómov sa dnes operuje endoskopicky endonazálne. Kraniotómia je indikovaná len u obrovských invazívnych tumorov a malígnych prolaktinómov. Je možný aj kombinovaný operačný prístup.
Nevýhodami chirurgickej liečby je možná recidíva, ktorá je popisovaná u 12 % mikroprolaktinómov a 50 % makroprolaktinómov. Komplikáciami môžu byť hypopituitarizmus s nutnosťou substitučnej liečby, poruchy zraku, diabetes insipidus, včasná likvorea, meningitída, stavy po transkraniálnych operáciách u gigantických adenómov a vzácne aj epilepsia a úmrtie.
3. Rádiologická liečba
Je indikovaná pri zlyhaní medikamentóznej a chirurgickej liečby, najmä pri veľkých invazívnych tumoroch a malígnych prolaktinómoch.
Pri veľkých reziduálnych tumoroch po operácii sa aplikuje konvenčná rádioterapia, alebo sa aplikuje stereotaktická rádioterapia – gamma nôž, fokálna radiačná liečba. Nevýhodou rádioterapie je neskorý nástup účinku, panhypopituitarizmus, poškodenie n. opticus, hypotalamu, rádionekróza mozgového tkaniva, zvýšené riziko cievnych mozgových príhod a sekundárnych nádorov mozgu.
4. Kombinovaná liečba
Indikácie k liečbe prolaktinómov sú útlakové príznaky tumoru a následky hyperprolaktinémie (hypogonadizmus, amenorea, oligomenorea, infertilita, impotencia, osteoporóza, osteopénia).
Indikácie k liečbe prolaktinómov sa zvažujú individuálne – podľa pohlavia, veku, snahy o graviditu. Len 5 – 10 % mikroprolaktinómov progreduje v raste, 30 % regreduje bez liečby, 24 % spontánne vymizne v gravidite s normalizáciou prolaktínu po pôrode, 25 – 45 % regreduje v menopauze s následnou úpravou, u 20 % mikroprolaktinómov dochádza po ukončení 2-ročnej liečby k trvalej úprave prolaktínu.
Ukončenie liečby je po 2 – 5 rokoch možné len pri normálnej hodnote PRL a úplnom vymiznutí tumoru a ak pri minimálnej dávke agonistu dopamínu nedochádza k vzostupu PRL ani rastu tumoru. Po vynechaní liečby je najvyšší počet recidív počas prvého roka.
Malígne prolaktinómy sa líšia od invazívneho makroadenómu len agresívnym správaním a tvorbou metastáz. Podávanie agonistov dopamínu tu vedie k redukcii masy tumoru, ale bez efektu na MTS. U malígnych prolaktinómov je jedinou dostupnou liečbou operácia s redukciou masy tumoru a zmenšením kompresie a rádioterapia s následnou chemoterapiou temozolomidom. Chemoterapia sa niekedy odporúča aj predoperačne.
Osobitným problémom je liečba prolaktinómov v gravidite, kde riziko symptomatického rastu prolaktinómu v dôsledku prerušenia liečby agonistami dopamínu a stimulujúceho vplyvu estrogénov je u mikroprolaktinómov 2,7 %, u makroprolaktinómov bez predchádzajúcej ablatívnej liečby 22,9 %, a u makroprolaktinómov s predchádzajúcu ablatívnou liečbou 4,8 %. Pri plánovaní gravidity u žien liečených agonistami dopamínu sa odporúča mechanická antikoncepcia až do nástupu pravidelného menštruačného cyklu.
Hypersekrécia somatotropínu (GH – growth hormone) a jeho periférneho mediátora IGF-1 (insulin-like-growth factor 1, inzulínu podobný rastový faktor 1) vedie k akromegálii alebo gigantizmu. Klinický obraz ochorenia závisí od veku, v ktorom hypersomatotropizmus nastal. V dospelosti sa prejavuje akromegáliou, v prepubertálnom veku gigantizmom, ktorý je veľmi zriedkavou endokrinopatiou.
Akromegália teda vzniká po ukončení telesného rastu – po uzatvorení rastových štrbín kosti nemôžu rásť lineárne, ale iba enchondrálne a apozíciou do šírky. Zväčšujú sa aj mäkké tkanivá a vnútorné orgány. Najnápadnejšími zmenami sú zhrubnuté a zväčšené akrálne časti tela – prsty, dolná čeľusť, zuby, pery, nos.
Príčiny akromegálie sú:
- primárny adenóm hypofýzy produkujúci GH
– mikroadenóm, makroadenóm, výnimočne karcinóm,
– MEN 1 (mnohopočetná endokrinná neoplázia 1), - hypotalamická dysregulácia s hypersekréciou GH
– hyperplázia somatotrofov s následnou adenomatóznou prestavbou, - paraneoplastický hypersomatotropizmus
– ektopická nádorová produkcia GH-RH alebo GH (nádory pankreasu, bronchov, karcinoid, feochromocytóm),
– eutopická nádorová produkcia GH-RH (hypotalamické gliómy, angiocytómy, hamartómy.
V čase rozvinutých klinických prejavov a diagnostikovania akromegálie sú somatotrofné adenómy hypofýzy obyčajne väčšie než 1 cm, mikroadenómy sa diagnostikujú asi u 20 % chorých. U štvrtiny až tretiny pacientov sa hypersekrécia GH spája aj s hypersekréciou PRL. Akromegália vzniká najčastejšie medzi 30. – 50. rokom života a postihuje približne rovnako mužov i ženy.
Prejavmi nadbytku GH sú – okrem najčastejšie sa vyskytujúcich zhrubnutých akier kože a podkožia – aj zvýšené potenie (88 – 91 % pacientov), spavosť, unaviteľnosť a letargia (42 – 82 %), artralgie (69 – 76 %), prírastok na hmotnosti (39 – 76 %), syndróm karpálneho tunela (46 – 70 %). Ďalšími prejavmi môžu byť kožné fibrómy a pigmentácie, artériová hypertenzia, struma či kardiomegália. Z iných endokrinných porúch je to prítomnosť hypogonadizmu, hyperprolaktinémie – galaktorea, hypokorticizmus.
Klinický obraz sa u chorých rozvíja postupne v priebehu viacerých rokov a postihnutý i okolie si vznikajúce zmeny nemusia dlho všímať. Na vyšetrenie prichádzajú pacienti väčšinou v pokročilom štádiu choroby s výrazne vyznačenou kombináciou uvedených príznakov. V pokročilom štádiu akromegálie sú už prítomné ireverzibilné poškodenia, predovšetkým kostné zmeny, kardiomyopatia, postihnutie optických nervov, výrazná defigurácia tváre. V pokročilom štádiu akromegálie je aj adenóm obyčajne veľký, a tým je aj znížená nádej na jeho úplné chirurgické odstránenie. Pre úspešnú terapiu je preto dôležité zachytiť akromegalický proces v počiatočnom štádiu.
Okrem laboratórnej diagnostiky (zvýšená bazálna hodnota IGF-1 v plazme a non-supresibilita GH pri oGTT) má nezastupiteľné miesto vizualizačná diagnostika – MR mozgu a oftalmologické vyšetrenie perimetra.
Osobitným variantom je akromegália tvoriaca súčasť mnohopočetnej endokrinnej neoplázie typu 1. Klinické prejavy akromegálie sa potom prekrývajú s príznakmi hyperparatyreózy, prípadne aj s príznakmi inzulinómu či gastrinómu.
U časti chorých s hyperpláziou somatotrofov možno predpokladať ako prvotnú príčinu hypersomatotropizmu hypotalamovú dysreguláciu. Podozrenie na paraneoplastickú formu akromegálie vzniká pri kombinácii klinicky a laboratórne zjavného hypersomatotropizmu s nenájdeným adenómom hypofýzy pri MR vyšetrení mozgu.
Terapia akromegálie:
- neurochirurgické odstránenie adenómu,
- iradiácia hypofýzy – lineárny urýchľovač, Leksellov gamma nôž – efekt liečby sa dostavuje do 2 rokov od ožiarenia; táto latentná perióda sa preklenuje medikamentóznou terapiou,
- farmakologická liečba (dopaminergní agonisti – bromokryptín, tergurid, syntetické analógy somatostatínu – oktreotid LAR, lanreotid LAR, lanreotid Autogel, antagonisti receptorov GH – somavert).
Cieľom terapie je odstrániť a deštruovať nádor hypofýzy. Zmierniť morbiditu sa u veľkého počtu chorých dosiahne len kombináciou uvedených terapeutických postupov.
Hypersekrécia kortikotropínu v adenómoch hypofýzy je príčinou centrálnej formy Cushingovho syndrómu.
Hypersekrécia tyreotropínu je zriedkavá forma hyperpituitarizmu a veľmi vzácna príčina centrálnej tyreotoxikózy. Na hypersekréciu TSH upozorňuje kombinácia tyreotoxikózy so zvýšenými koncentráciami TSH v plazme. Diagnózu potvrdí nález adenómu hypofýzy pri vizualizačnom vyšetrení. Prvým terapeutickým krokom je odstránenie adenómu hypofýzy. Ak sa remisia tyreotoxikózy nedosiahne, nasleduje tyreoidektómia alebo liečba rádiojódom.
Hypersekrécia gonadotropínov je ďalšou zriedkavou formou hyperpituitarizmu. Môže ísť o nadprodukciu FSH aj LH alebo o hypersekréciu jedného z nich. Klinickým následkom ich nadprodukcie je u žien oligomenorea, u mužov impotencia. Laboratórne nachádzame v plazme kombináciu subnormálnych hodnôt testosterónu alebo estradiolu s vysokými hodnotami gonadotropínov – teda rovnaká konštelácia ako pri periférnom hypogonadizme. Na autonómnu nadprodukciu gonadotropínov upozorňuje non-supresibilita ich plazmatickej hladiny pri podávaní sexuálnych steroidov, diagnózu ozrejmí aj nález adenómu hypofýzy. Terapiou je odstránenie adenómu hypofýzy neurochirurgicky.
Okrem uvedených symptomatických foriem hyperpituitarizmu existuje aj bezpríznaková hypersekrécia FSH, LH, TSH a alfa-podjednotky glykoproteínových tropínov. Zdá sa, že väčšina hypofýzových makroadenómov, ktoré sa klinicky javia ako sekrečne inaktívne, vytvára a vydáva do obehu niektorý z uvedených glykoproteínových tropínov, ich kombinácie alebo samostatnú alfa-podjednotku. Príčina bezpríznakovosti pri hypersekrécii TSH, FSH a LH nie je známa, azda ide o biologicky inaktívny, avšak imunologicky reagujúci materiál. Terapia je neurochirurgická a je indikovaná pre lokálny mechanický syndróm.
Kombinovaná hypersekrécia pri symptomatických hyperpituitárnych stavoch pozostáva najčastejšie z pridruženia nadmernej tvorby PRL ku klinicky dominujúcej hypersekrécii GH alebo ACTH. Nadmerná tvorba alfa-podjednotky sa môže spájať s hypersekréciou gonadotropínov, prolaktínu alebo GH.
Štítna žľaza

Strumy možno rozdeliť podľa rôznych kritérií na:
- difúzne a uzlové (morfologické hľadisko),
- parenchýmové a koloidné (histologické hľadisko),
- eufunkčné, hypofunkčné a hyperfunkčné (funkčné hľadisko),
- benígne a malígne (onkologické hľadisko),
- endemické a sporadické (epidemiologické hľadisko).
Eufunkčná struma je difúzne alebo uzlovité zväčšenie štítnej žľazy s normálnou funkciou, ktoré nie je spôsobené zápalovým, autoimunitným ani malígnym procesom (synonymum – netoxická struma, „struma simplex“).
Difúzna struma sa definuje ako každé zrakom alebo hmatom zistené rovnomerné zväčšenie štítnej žľazy, ktorej laterálne laloky (pravý a ľavý) sú objemnejšie ako terminálny článok palca vyšetrovanej osoby. Prítomnosť uzla znamená vždy strumu, bez ohľadu na jej veľkosť. V krajinách s normalizovaným príjmom jódu, ku ktorým patrí aj Slovensko, sú objemy štítnej žľazy malé a netoxická difúzna struma je z epidemiologického, ako aj klinického hľadiska nevýznamná. V súvislosti s výskytom klinicky manifestnej uzlovej strumy v populácii sa uvádza 4 – 7 % prípadov. USG vyšetrenia, ktoré majú nezastupiteľné a prioritné miesto v diagnostike ochorení štítnej žľazy, odhaľujú uzlové zmeny, a to aj drobné a klinicky nevýznamné až v 15 – 40 % dospelej populácie, autopsie až v 30 – 60 % prípadov.
Príčiny uzlovej strumy sú v porovnaní s minulosťou diametrálne odlišné. Väčšinu uzlov (približne 60 %) predstavujú folikulárne adenómy. Relatívny výskyt karcinómov v uzloch dosahuje 5 – 10 %. Podiel hyperplastických uzlov a viacuzlovej strumy, ktoré boli hlavným patologicko-anatomickým nálezom v podmienkach jódového deficitu, poklesol približne na 20 %. Zvyšok tvoria cysty, posthemoragické pseudocysty, tyreoiditídy a granulomatózne ochorenia.
Pri vzniku netoxickej uzlovej strumy (adenomatózneho uzla a viacuzlovej strumy) hrajú dôležitú úlohu geneticky dané rozdielne vlastnosti folikulárnych buniek.
S poklesom absolútneho počtu uzlových strúm v krajinách s normalizovaným príjmom jódu stúpa relatívny podiel tyreoidálnych neoplázií – adenónov a karcinómov v existujúcich sporadických uzlových strumách. Preto je v súčasnosti potrebné pristupovať ku každej uzlovej strume ako k potenciálnej malignite. Donedávna rozšírený názor, že v multinodóznej strume je oveľa menšia pravdepodobnosť malignity než v solitárnom uzle, už neplatí. Pacienti s viacuzlovou strumou majú väčšie riziko malignity než pacienti so solitárnym uzlom.
Skupinu malígnych nádorov štítnej žľazy s najväčším výskytom a klinickým významom – t. j. primárne malígne epitelové nádory – predstavujú nádory vychádzajúce z folikulárnych buniek štítnej žľazy (papilárny, folikulárny, anaplastický karcinóm) a nádory vychádzajúce z parafolikulárnych buniek (medulárny karcinóm). Napriek svojej vzácnosti tvoria nezanedbateľnú skupinu nádorov ŠŽ malígne lymfómy, predstavujú menej ako 5 % jej neoplázií. V štítnej žľaze môžu byť prítomné aj MTS, najmä melanómu, karcinómu obličky, prsníka, pľúc, GIT-u, tiež sarkómy pochádzajúce z buniek väziva.
Väčšina uzlov v štítnej žľaze, či už malígnej alebo benígnej povahy, je dlho asymptomatická. Po rokoch nediagnostikované uzly môžu spôsobiť mechanický syndróm, deglutinačné a dýchacie ťažkosti. Malígny uzol sa vo veľkej väčšine prípadov dlho, často aj niekoľko rokov, klinicky ničím nelíši od benígnej uzlovej strumy. Pre diferenciálnu diagnostiku uzlovej strumy, a najmä pre rozhodnutie o operačnom riešení (strumektómia, lobektómia), je rozhodujúcim krokom tenkoihlová biopsia s cytologickým vyšetrením.
Tenkoihlová biopsia (FNAB) sa má uskutočňovať pod USG kontrolou. Donedávna sa kládol dôraz na vykonanie biopsie z dominantného uzla (uzlov), v súčasnosti sa pri výbere uzla na biopsiu uprednostňujú skôr USG kritériá, ktoré poukazujú na možnosť malignity v uzle (solídny charakter uzla s hypoechogenitou a nehomogénnou echoštruktúrou tkaniva, neostré zvlnené okraje uzla, chýbanie „halo“ lemu, prítomnosť psamómov, centrálne prekrvenie pri dopplerovskom vyšetrení).
Väčšina cytológov triedi výsledok cytologického vyšetrenia do 5 kategórií – malígny, suspektný, atypický alebo neurčitý, benígny a nedostatočný (nereprezentatívny). Pri nedostatočnom odbere materiálu na cytologické vyšetrenie je potrebné biopsiu zopakovať. Výsledok cytologického vyšetrenia – prvé 3 kategórie – malígny, suspektný a atypický – vyžadujú operačné riešenie a histologické potvrdenie diagnózy. Ak je cytologický výsledok benígny, potvrdí sa diagnóza netoxickej uzlovej strumy a postupuje sa konzervatívne, pokiaľ nie sú splnené kritéria na operáciu. Význam cytológie spočíva nielen vo vyselektovaní pacientov indikovaných na operáciu, ale súčasne aj v „ochrane“ pacienta pred operáciou, ktorá nebola nevyhnutná. Cytologické vyšetrenie má vysokú pozitívnu prediktívnu hodnotu v prípadoch papilárneho, medulárneho, anaplastického a metastatického karcinómu. Slabinou cytológie je, že sa nedá rozlíšiť folikulárny adenóm od folikulárneho karcinómu a folikulárneho variantu papilárneho karcinómu. Problematické je aj hodnotenie pseudocysticky degenerovaných uzlov. Aspiračná cytodiagnostika je v prípade cýst a pseudocýst aj liečebnou metódou.
Karcinóm štítnej žľazy je pomerne zriedkavý, tvorí približne 1,1 – 1,9 % malígnych nádorov. Je však najčastejším karcinómom endokrinného systému (viac než 90 %). Ženy sú postihnuté trikrát častejšie než muži. U žien patrí do prvej desiatky najčastejšie sa vyskytujúcich malígnych nádorov, u detí a adolescentov je piatou najčastejšou malignitou. Má dobrú prognózu, na čom sa podieľa najmä epidemiologicky najvýznamnejší diferencovaný karcinóm. Zomiera naň 6 % pacientov. Incidencia karcinómu štítnej žľazy za posledné 3 dekády stúpla celosvetovo aj na Slovensku (podiel rádioaktívneho spádu pri testovaní atómových bômb a pri haváriách jadrových reaktorov, znečisťovanie životného prostredia, ale najmä zlepšená diagnostika predovšetkým malých karcinómov). V súčasnosti čelíme „epidémii“ papilárneho mikrokarcinómu. Karcinóm štítnej žľazy sa vo väčšine prípadov prejavuje ako uzlová struma.
Varianty diferencovaného karcinómu štítnej žľazy:
Etiologicky sú všetky typy karcinómu štítnej žľazy spôsobené abnormalitami génov riadiacich rast a proliferáciu buniek. Môže ísť o mutácie tumor-supresných génov, aktiváciu onkogénov, poruchu reparačných mechanizmov DNA alebo ich kombináciu. Jedným z najdôležitejších faktorov, ktoré spôsobujú poškodenie génov, je radiácia. Genetické poruchy sú najlepšie preskúmané na familiárnej forme medulárneho karcinómu. Jeho príčinou je germinatívna mutácia RET protoonkogénu.
Liečba diferencovaného karcinómu spočíva v troch pilieroch:
- operačný výkon,
- liečba rádiojódom,
- a dlhodobá supresívna a substitučná liečba tyreoidálnymi hormónmi.
Chirurgická liečba predstavuje najdôležitejšiu terapeutickú modalitu. Dôkladne vykonaná totálna tyreoidektómia s disekciou centrálneho – a podľa potreby aj laterálneho – kompartmentu vytvára podmienky pre úspech liečby rádiojódom, pre minimalizáciu recidív a normálne prežívanie pacientov. Operácie karcinómov štítnej žľazy, a najmä reoperácie (dokončenie totálnej tyreoidektómie s disekciou krčných kompartmentov), musí vykonávať erudovaný chirurg na pracovisku s dostatočnými skúsenosťami s týmto typom operácií.
Liečba rádiojódom (131I) sa podáva na špecializovaných lôžkových oddeleniach nukleárnej medicíny. Môže sa začať najskôr 6 týždňov po operácii a reoperácii, ak akumulácia po podaní malej diagnostickej dávky nie je vyššia ako 10 %. Pred podaním rádiojódu majú pacienti dodržiavať aspoň 2 mesiace nízko jódovú diétu (nekonzumovať morské ryby, niektoré minerálky, multivitamíny s obsahom jódu, dezinfekčné jódové roztoky). Aby bola tyreoeliminácia rádiojódom efektívna, je potrebné dosiahnuť dostatočnú stimuláciu reziduí tyreotropínom (TSH). Koncentrácia TSH v sére pred podaním tyreoeliminačnej dávky 131I má dosiahnuť minimálne 30 mU/l – hlboká hypotyreóza navodená (endogénna stimulácia) 6 – 8 týždňov po operácii. Stimuláciu možno prípadne dosiahnuť podaním rekombinantného ľudského TSH (rhTSH) – tu sa liečba tyroxínom nevysadzuje a pacienti nemajú symptómy hypotyreózy. Prvé liečebné podanie 131I po operácii sa nazýva tyreoeliminácia alebo ablácia, pričom jej úlohou je deštruovať akékoľvek zvyškové tyreoidálne tkanivo v lôžku štítnej žľazy. Ide najmä o likvidáciu zvyškov normálneho tkaniva, ktoré má oveľa vyššiu akumulačnú schopnosť než nádorové tkanivo. 131I umožňuje odhaliť predtým nediagnostikované MTS, ablácia reziduálneho normálneho tyreoidálneho tkaniva umožňuje včasné detegovanie recidívy pomocou tyreoglobulínu – prípadne celotelovej scintigrafie – a umožňuje jej liečbu 131I. Hoci je podávanie rádiojódu bezpečnou terapeutickou metódou, zriedka sa môžu vyskytnúť včasné alebo neskoré komplikácie. Riziko ich vzniku koreluje s veľkosťou individuálnej a kumulatívnej dávky. Absolútnymi kontraindikáciami liečby rádiojódom sú gravidita a laktácia. Relatívnymi sú chronická renálna insuficiencia v štádiu dialyzačnej liečby a leukopénia.
Po liečbe rádiojódom nastupuje supresívna liečba, ktorej cieľom je po iniciálnej úprave hypotyreózy zabrániť TSH dependentnému rastu reziduálneho tumoru. V liečbe sa používa levotyroxín.
V dlhodobom sledovaní pacientov sa sleduje koncentrácia bazálneho a stimulovaného tyreoglobulínu v sére, USG krku, poterapeutická (veľká dávka) alebo diagnostická (malá dávka) celotelová scintigrafia s rádiojódom. U pacientov s MTS (aj s predpokladanými), ktoré nevychytávajú rádiojód, sa dopĺňa CT vyšetrenie. Zatiaľ čo USG vyšetrenie je predoperačne cennou a najviac využívanou diagnostickou metódou, pooperačne jej význam klesá. „Negatívny“ USG nález nemusí znamenať remisiu ochorenia. Perzistujúce malígne tkanivo alebo recidíva môžu byť lokalizované za tracheou, ezofágom, v hlbokých štruktúrach krku, kde USG diagnostika zlyháva. Okrem diagnostiky CT a MR možno využiť aj PET, scintigrafiu s 99mTc-sestamibi a scintigrafiu s 99mTc-tetrofosmínom.
Najcennejším markerom v dlhodobom sledovaní pacientov s diferencovaným karcinómom štítnej žľazy je tyreoglobulín (TG). Za najspoľahlivejšiu metódu sledovania pacientov sa v súčasnosti považuje kombinácia vyšetrenia TG po stimulácii rhTSH (rhTSH-TG) a USG krku. Diagnostická alebo terapeutická scintigrafia sa indikuje pri vzostupe stimulovaného TG.
Externá rádioterapia, chemoterapia a biologická liečba sa podávajú zriedka.
Jedinou liečebnou modalitou medulárneho karcinómu štítnej žľazy je dostatočne radikálny chirurgický výkon vo včasnom štádiu ochorenia. V prípade vzdialených metastáz je choroba inkurabilná a všetky terapeutické postupy majú iba paliatívny charakter. V prípade medulárneho karcinómu je dôležitá predoperačná diagnostika – pri určovaní rozsahu ochorenia patrí z biochemických vyšetrení prvé miesto kalcitonínu, CEA a vápniku. V prípade predoperačne dokázaného medulárneho karcinómu sa má vykonať aj genetické vyšetrenie. U nositeľov mutovaného RET onkogénu sa má pátrať po prítomnosti feochromocytómu, pričom pri jeho dôkaze musí extirpácia feochromocytómu predchádzať operácii štítnej žľazy.
Aj v pooperačnom stagingu patrí rozhodujúce miesto onkomarkerom – kalcitonínu a CEA. Prognóza pacientov s medulárnym karcinómom štítnej žľazy je horšia ako u pacientov s diferencovaným karcinómom štítnej žľazy. Medulárny karcinóm má oveľa väčší metastatický potenciál.
Anaplastický karcinóm štítnej žľazy má rýchlu progresiu, metastazovanie a veľmi zlú prognózu, preto sú terapeutické možnosti limitované – snahou je čo najradikálnejší chirurgický výkon.
U pacientov s karcinómom štítnej žľazy je nutná doživotná dispenzarizácia.
Prištítne telieska
Ich najčastejším ochorením je primárna hyperparatyreóza definovaná ako generalizovaná porucha kalciového, fosfátového a kostného metabolizmu, ktorá je dôsledkom dlhodobej zvýšenej sekrécie parathormónu a je vyvolaná primárne poruchou funkcie prištítnych teliesok. Ide buď o sporadickú formu (90 %), alebo familiárnu formu (10 %), napríklad ako súčasť MEN 1, MEN 2A, familiárnej izolovanej hyperparatyreózy, syndrómu hyperparatyreóza – čeľusť (jaw tumor syndrome spojeného s osifikujúcim fibroseróznym tumorom čeľuste, cystami a hamartómami obličiek a Wilmsovým tumorom).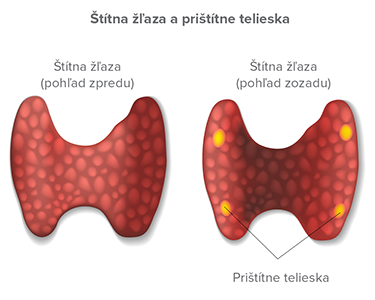
Etiológia:
- adenóm prištítneho telieska solitárny, zriedka mnohopočetný (80 – 85 %),
- primárna hyperplázia prištítnych teliesok (10 – 15 %),
- karcinóm prištítneho telieska (vzácne).
Až v 80 % je primárna hyperparatyreóza asymptomatická.
Symptomatická hyperparatyreóza:
- kostné zmeny – hyperparatyreózna osteodystrofia – bolesti kĺbov, chrbtice, subperiostálne rezorpcie až akroosteolýza, kostné cysty, tzv. hnedé tumory, osteoporóza hlavne kortikálnej kosti, patologické fraktúry, deformity,
- renálne postihnutie – nefrolitiáza, recidivujúce uroinfekcie, nefrokalcinóza, polyúria, polydipsia,
- GIT symptómy – anorexia, nauzea, zvracanie, bolesti brucha, chudnutie, vredová choroba žalúdka, pankreatitída,
- kardiovaskulárne symptómy – artériová hypertenzia, poruchy srdcového rytmu,
- neuromuskulárne symptómy – únava, svalová slabosť, parestézie, symetrické postihnutie proximálneho svalstva s poruchami chôdze, hyperreflexiou a EMG zmenami, únava, apatia, depresia, dezorientácia až kóma.
- očné príznaky – prúžkovité kalcifikácie v rohovke a Ca depozity v spojivkovom vaku.
Diagnostika hyperparatyreózy je:
- laboratórna – hyperkalciémia, zvýšený nesuprimovaný parathormón, ďalej sa vyšetruje kalciúria, fosfatémia, kalcidiol, kostný izoenzým alkalickej fosfatázy,
- lokalizačná – USG – senzitivita 69 – 96 % pri adenóme, pri hyperplázii je prínos USG malý,
- scintigrafia
– dvojfázová scintigrafia 99mTcMIBI (adenóm),
– subtrakčná 99mTcMIBI alebo 99mTc-tetrofosmín, - SPECT/CT scintigrafia,
- MR/CT vyšetrenie,
- tenkoihlová biopsia so stanovením PTH v aspiráte,
- venózna katetrizácia so stanovením PTH.
Liečba primárnej hyperparatyreózy je chirurgická, extirpácia adenómu/adenómov, pri hyperplázii subtotálna paratyreoidektómia (odstránenie 3,5 teliesok) alebo totálna paratyreoidektómia s ich autotransplantáciou.
Kritériom na chirurgickú liečbu je symptomatická hyperparatyreóza. V prípade, že ide o asymptomatickú hyperparatyreózu, sa operačné riešenie volí pri pacientoch do 50 rokov, kalciémii viac ako 0,25 mmol/l nad hornou hranicou referenčného rozsahu, pri renálnej insuficiencii bez inej preukázateľnej príčiny či poklese kostnej denzity. Úskaliami neúspešnej chirurgickej liečby môže byť zámena adenómu prištítneho telieska za inú štruktúru, atypická lokalizácia adenómu alebo mnohopočetný adenóm.
Medikamentózna liečba primárnej hyperparatyreózy je indikovaná pri kontraindikácii chirurgického riešenia, prípadne ak pacient operáciu odmietne.
Nadobličky

- Cushingov syndróm – v dôsledku zvýšenej sekrécie kortizolu (50,4 %),
- primárny hyperaldosteronizmus ako následok autonómnej hypersekrécie aldosterónu (38,2 %),
- adrenálna virilizácia z nadmernej sekrécie nadobličkových androgénov (10,2 %),
- adrenálna feminizácia z nadmernej sekrécie nadobličkových estrogénov (1,2 %).
Adrenokortikálna hypersekrécia vedúca k týmto syndrómom býva selektívna len pri primárnom hyperaldosteronizme, kým hypersekrécia kortizolu alebo nadobličkových estrogénov sa často spája s nadbytočnou tvorbou nadobličkových androgénov. Klinický obraz závisí od dominantne vytváraného steroidu. Ku kombinácii všetkých štyroch sekrečných syndrómov môžu viesť niektoré karcinómy kôry nadobličiek. Morfologickým podkladom je bilaterálna hyperplázia, adenóm alebo karcinóm nadobličky. Výnimkou je zriedkavá adrenálna feminizácia, zdrojom ktorej sú neoplázie, zvyčajne karcinóm adrenokortexu.
Cushingov syndróm (CS)
Ochorenie zapríčinené hypersekréciou kortizolu z ktorejkoľvek etiologickej príčiny.
Etiológia:
- ACTH-dependentný CS (80 %)– adenóm hypofýzy:
– ektopická sekrécia ACTH,
– ektopická sekrécia CRH, - ACTH-independentný CS (19 %) – adenóm nadobličky:
– karcinóm nadobličky,
– bilaterálna (makro/mikro) nodulárna hyperplázia nadobličiek, - iatrogénny CS – klinický obraz spôsobený dlhodobým podávaním vysokých dávok kortikoidov.
Klinický obraz CS nemusí byť typický – od oligosymptomatickej a atypickej formy až po plne rozvinutý CS – centripetálny typ obezity, hypotrofia koreňového svalstva, facies lunata, plethora tváre, cervikálny tukový hrboľ, fialové strie, hirzutizmus, akné, svalová slabosť, psychická alterácia, impotencia, oligo/amenorea. Sprievodným príznakmi sú artériová hypertenzia (nad 50 %), diabetes mellitus alebo porucha glukózovej tolerancie (30 %), vredová choroba duodena, glaukóm.
V diagnostike má okrem základného laboratórneho skríningu (polyglobúlia, lymfopénia, neutropénia, hyperglykémia, hypokaliémia, dyslipidémia) rozhodujúci význam laboratórne endokrinologické vyšetrenie – vysoké hladiny voľného močového kortizolu/24 hod., narušený cirkadiánny rytmus v diurnálnom profile – bez nočnej supresie, ACTH. Nízke hladiny v sére svedčia pre periférnu formu CS, vysoké alebo normálne hladiny pre centrálnu formu CS, veľmi vysoké pre paraneoplastický CS. Za účelom ďalšej diferenciálnej diagnostiky sa realizujú u pacientov funkčné testy (nočný dexametazónový test, slabšia a silnejšia dexametazónová blokáda, CRH test). Zo zobrazovacích metód sa využíva MR hypofýzy, CT nadobličiek, sampling sinus petrosus inferior – selektívne stanovenie ACTH u centrálnej formy CS, počítačový perimeter.
Cieľom liečby CS je odstránenie príčiny hyperkorticizmu, stratégia liečby sa líši od patogenetickej formy. Liečba centrálnej formy CS je chirurgická – transsfenoidálna adenomektómia. Totálna bilaterálna adrenalektómia je v prípade zlyhania neurochirurgického zákroku na hypofýze a neefektívnej rádioterapie jednou z posledných možností liečby centrálneho CS. Pri recidíve CS je vhodnou metódou voľby aj rádioterapia – lineárny urýchľovač či Leksellov gamma nôž. Jediným špecifickým farmakologickým prostriedkom na centrálnu formu CS je pasireotid (Signifor) – je indikovaný, ak chirurgický zákrok nie je možný , alebo ak chirurgická liečba zlyhala.
Liečba periférnej formy CS – chirurgická liečba – adrenalektómia (odstránenie adenómu nadobličky). Uskutočňuje sa na špecializovaných urologických, prípadne chirurgických pracoviskách – prevažne laparoskopicky. Peroperačné a pooperačné zabezpečenie pacienta hydrokortizónom je nevyhnutnosťou! Pri karcinóme nadobličky nasleduje po adrenalektómii chemoterapia a/alebo liečba adrenolytikom, respektíve adrenostatickými prípravkami.
Pri paraneoplastickom CS je optimálnou terapiou odstránenie nádoru so sekréciou ACTH alebo CRH. V prípade inkompletného odstránenia je potrebná chemoterapia, aktinoterapia a paliatívna farmakologická liečba. Vo výnimočných prípadoch sú účinné analógy somatostatínu.
Primárny hyperaldosteronizmus
Je definovaný ako nadmerná a relatívne autonómna produkcia aldosterónu. Vymyká sa z regulácie renín-angiotenzínového systému, ktorý je suprimovaný. U časti chorých je prítomná hypokaliémia, zriedkavo sú pacienti normotenzní a môžu mať normálne hodnoty plazmatického a močového aldosterónu.
Adenómy produkujúce aldosterón (APA) sa v porovnaní s idiopatickým hyperaldosteronizmom (IHA) vyskytujú u mladších pacientov. Adenómy sú častejšie u žien. Prevalencia v populácii hypertonikov je 5 – 10 % a asi 1,5 % v bežnej populácii.
V diagnostike – okrem anamnézy – fyzikálneho vyšetrenia (artériová hypertenzia – najmä hypertonici s hypokaliémiou, rezistentní na liečbu, závažná diastolická hypertenzia, hypertonici v mladom veku) majú nezastupiteľné miesto aj odbery plazmatickej renínovej aktivity (PRA) alebo plazmatickej koncentrácie renínu (PRC), plazmatická hladina aldosterónu a aldosterónovo-renínový pomer (ARR). Pred vyšetrením týchto parametrov je u pacienta nutné upraviť kaliémiu, vynechať antihypertezíva – beta-blokátory, diuretiká, ACEI, AT II blokátory a dihydropyridíny. Pred testovaním možno v liečbe ponechať prazosín alebo iné alfa-blokátory, hydralazíny a retardovaný verapamil.
V rámci ďalšej diferenciálnej diagnostiky sa realizujú dynamické testy, a to buď supresívne (testy so soľnou záťažou, kaptoprilový test), alebo stimulačné (vplyv polohy a času, infúzia angiotenzínu II, odpoveď na dexametazon). Pri dif. diagnostike primárneho hyperaldosteronizmu sa využíva aj stanovenie deoxykortikosterónu (DOC), 18-OH-DOC, kortikosterónu, 18-OH- kortikosterónu, 18-OH kortizolu a 18-oxokortizolu.
Lokalizácia aldosterónovej nadprodukcie sa realizuje CT alebo MR vyšetrením, tiež sa využíva adrenálna venografia a separovaný odber z adrenálnych vén (ACS).
Liečba PH je chirurgická – unilaterálna adrenalektómia, predoperačne je však nutné pacienta pripraviť podávaním spironolaktónu. Pri aldosterón produkujúcich karcinómoch nadobličiek je liečbou voľby Trilostan – inhibítor 3-beta-HSD a tiež adrenolytikum mitotan.
Feochromocytóm
Zriedkavý nádor vychádzajúci z drene nadobličiek, ktorý produkuje obvykle veľké množstvo katecholamínov (adrenalín, noradrenalín), a tým vedie k závažnému zvýšeniu tlaku krvi. Okrem drene nadobličiek môže vychádzať aj z ganglií uložených v brušnej alebo hrudnej dutine, vtedy sa označuje ako paraganglióm. Aj keď sa feochromocytóm vyskytuje veľmi zriedkavo, svojimi následkami môže pacienta ohroziť na živote. Vyskytuje sa asi u 1 % ľudí s artériovou hypertenziou a až 24 % feochromocytómov je vrodených, zriedka sú zhubné.
Okrem artériovej hypertenzie nemusia mať pacienti žiadne iné príznaky, avšak pri náhlom vyplavení hormónov sa u nich môže vyskytnúť náhla cefalea, tachykardia, potenie, pacient je ohrozený vznikom malígnej arytmie, cievnej mozgovej príhody či infarktu myokardu.
Z laboratórnej diagnostiky sa stanovujú voľné metanefríny v plazme (senzitivita 99 %), CT vyšetrenie v diagnostike má senzitivitu približne 95 %, avšak špecificita je len cca 70 % – vyššiu špecificitu má MR vyšetrenie.
Funkčné vyšetrenie – MIBG (metajódbenzylguanidín) – sa realizuje pri podozrení na disemináciu a neobvyklú lokalizáciu feochromocytómu. Rovnako PET vyšetrenie sa využíva pri diagnostike malígneho feochromocytómu, feochromocytómu v neobvyklej lokalizácii a MTS feochromocytómu.
Dedičný feochromocytóm je spojený s mnohopočetnou endokrinnou neopláziou 2 (MEN 2a a MEN 2b), von Recklinghausenovou neurofibromatózou NF-1, von Hippel Lindau syndrómom (VHL) a familiárnym paragangliómom spojeným s mutáciou génov kódujúcich sukcinátdehydrogenázu.
Jediná liečba feochromocytómu je chirurgická. Veľmi dôležitá je predoperačná príprava zameraná na kontrolu artériovej hypertenzie, udržanie stabilizovaného TK (blokátor alfa-adrenergných receptorov) a minimalizovanie nežiaducich účinkov počas anestézie a operácie.
Adrenálne incidentalómy
Sú to atologické adrenálne štruktúry zistené náhodne pri zobrazení dutiny brušnej (USG, CT, MRI), ktoré bolo indikované z iných príčin, ako je podozrenie na adrenálnu patológiu. V súčasnosti sa pod pojmom adrenálny incidentalóm rozumie patologický útvar veľkosti 1 cm a viac. Definícia nezahŕňa incidentalómy s prejavmi endokrinnej nadprodukcie a incidentalómy u chorých so známou malignitou.
Je to najväčšia skupina nadobličkových tumorov. Ich počet v posledných rokoch pribúda vďaka častejšiemu používaniu stále dokonalejších zobrazovacích metód a so stúpajúcim počtom realizovaných vyšetrení. Vyskytujú sa častejšie u starších osôb, u žien, hypertonikov s esenciálnou hypertenziou, diabetikov a u pacientov s malignitami. Veľké CT štúdie uvádzajú prevalenciu do 2,5 %, v populácii nad 55 rokov až 4,4 %. Adrenálne incidentalómy delíme na hormonálne aktívne a neaktívne (afunkčné), podľa charakteru na benígne a malígne. Najčastejšie ide o adenómy, prevažne afunkčné, malígne lézie – adrenokortikálny karcinóm, MTS, lymfómy, cysty, myelolipómy, feochromocytóm, zriedkavejšie ganglioneurómy, angiolipóm a iné. Dôležité je otestovať hormonálnu aktivitu takto zachytených štruktúr, vylúčiť glukokortikoidnú, katecholamínovú, androgénnu a estrogénovú nadprodukciu, vylúčiť primárny hyperaldosteronizmus.
Diferenciálna diagnostika spočíva v odlíšení benígnych lézií od malígnych, v rozlíšení jednotlivých benígnych lézií na základe zobrazovacích vyšetrení a v odlíšení hormonálne aktívnych más od neaktívnych.
Jedinou kuratívnou liečbou je chirurgická terapia – adrenalektómia.
MEN syndróm
Syndrómy mnohonásobnej endokrinnej neoplázie sú ochorenia s familiárnym výskytom, pri ktorých sa u postihnutého jedinca vyskytujú súčasne alebo v rôznych obdobiach života hyperfunkčné nádory (často zhubné) alebo hyperplázie viacerých endokrinných žliaz.
Hormonálna produkcia nemusí mať klinický korelát. Súčasťou MEN môžu byť aj neendokrinné postihnutia.
MEN 1 – syndróm vzniká kombináciou viac než 20 endokrinných alebo neendokrinných tumorov, pričom sú prítomné aspoň dva z troch postihnutí (prištítne telieska, pituitárne a neuroendokrinné tumory GIT-u).
Endokrinné tumory – primárna hyperparatyreóza (95 %), neuroendokrinné tumory GIT-u (gastrinóm 40 %, inzulinóm 10 %, PP tumor 20 %, glukagonóm, VIPom), nádory hypofýzy, karcinoidné tumory, adrenokortikálne nefunkčné adenómy.
Neendokrinné tumory – lipómy (30 %), kolagenomatózy (70 %), angiofibrómy tváre, CNS tumory (meningeómy, ependymómy), leiomyómy.
MEN 2
MEN 2A – subtyp zahŕňa 80 % vrodených medulárnych karcinómov štítnej žľazy (MTC), feochromocytóm a hyperparatyreózu. Pri MEN 2A môže byť prítomná aj kožná lichenová amyloidóza a Hirschprungova choroba.
Familiárny medulárny karcinóm štítnej žľazy – FMTC je charakterizovaný výskytom iba MTC bez prítomnosti feochromocytómu a hyperparatyreózy aspoň u 4 príslušníkov rodiny s MTC.
MEN 2B – medulárny karcinóm štítnej žľazy (100 %), feochromocytóm (50 %) a typický klinický obraz (facies, marfanoidný habitus, očné abnormality, muskuloskeletárne prejavy, generalizovaná ganglioneuromatóza). V 90 % prípadov sú prítomné gastrointestinálne prejavy – bolesti brucha, obstipácia, intermitentné hnačky, megakolon, ktoré môžu byť dominujúce už v novorodeneckom období.
MEN 4 – asociácia tumorov prištítnych teliesok, adenohypofýzy, prípadne aj nadobličiek, obličiek a reprodukčných orgánov.
Neuroendokrinné tumory (NET)
Majú svoj pôvod v bunkách difúzneho neuroendokrinného systému (DNES). Tvoria extrémne heterogénnu skupinu, jednak vzhľadom na histologické a endokrinné vlastnosti, ďalej pre klinické prejavy ochorenia a v neposlednom rade aj pre tendenciu tvorby metastáz. Ich výskyt je pomerne nízky, pretože väčšina NET-ov je klinicky nemých a teda nezistených. Prevalencia je najvyššia v 5. – 6. dekáde života. Karcinoidy sa najčastejšie vyskytujú ako primárne nádory gastrointestinálneho traktu (67,5 %), môžu sa nachádzať aj v pľúcach (25,3 %), ováriách, testes, týme a v obličkách (7,2 %). Z pankreatických neuroendokrinných tumorov (PET) je najčastejší inzulinóm, ďalej gastrinóm, VIP-óm a glukagonóm. Ostatné funkčné PET-y sa vyskytujú veľmi raritne (somatostatinómy, nádory produkujúce ACTH). Nefunkčné nádory pankreasu sa podľa viacerých zdrojov podieľajú na incidencii PET 25 – 50 % .
Diagnostika NET-ov je založená na klinických prejavoch, ktoré sú často nešpecifické (kašeľ, hemoptýza, opakované bronchopneumónie, GIT ťažkosti, flush – záchvatovité začervenanie kože hlavy, krku a hrudníka a ďalšie) a na dôkaze hormonálnej produkcie nádoru. Dnes je známych takmer 100 peptidov produkovaných týmito nádormi – hormonálna nadprodukcia serotonínu, gastrínu, inzulínu, C-peptidu, kortizolu, ACTH, kalcitonínu, STH, somatostatínu, glukagónu, v moči 5-hydroxyindoloctová kyselina – metabolit serotonínu, ktorý je zvýšený u 75 % pacientov s karcinoidom. Z lokalizačnej diagnostiky primárneho nádoru v oblasti GIT-u sa využívajú endoskopické vyšetrenia – gastroskopia, kolonoskopia, rektoskopia, na diagnostiku v tenkom čreve sa využíva enteroklýza a v indikovaných prípadoch kapsulová endoskopia. Dôležitú úlohu v lokalizácii pankreatických NET-ov zohráva endoskopická ultrasonografia. RTG hrudníka a bronchoskopia sú indikované pri podozrení na bronchiálny karcinoid. USG a CT je indikované pri podozrení na MTS. Prelom v diagnostike NET-ov nastal pri zavedení octreoscanu, a to vzhľadom na prítomnosť receptorov somatostatínu vo viacerých neuroendokrinných tumoroch na povrchu nádorových buniek. Pri detekcii karcinoidných nádorov sa využíva aj pozitrónová emisná tomografia (PET).
Liečba NET-ov je chirurgická – resekcia primárneho nádoru a MTS (debulking pri nemožnosti radikálnej resekcie).
Medikamentózna liečba – indikovaná je bioterapia, teda podávanie analógov somatostatínu alebo interferónu alfa. Chemoterapia – efekt cytostatickej liečby je nízky, preto sa dnes indikuje len u anaplastických a málo diferencovaných neuroendokrinných karcinómov.
Rádionuklidová terapia (PPRT peptid receptor radionuclide therapy) – väčšina NET exprimuje somatostatínové receptory. To umožňuje nadviazať izotop ytria alebo litécia na oktreotid, rádiofarmakum sa vychytáva cielene v mieste nádoru. Liečba vedie k zlepšeniu u pacientov s pokročilým nádorom. Túto liečbu poskytuje len niekoľko pracovísk v Európe.
Symptomatická liečba sa využíva pri ovplyvnení príznakov zaťažujúcich pacienta, podávajú sa kortikoidy, H2 blokátory, chlórpromazín, antagonisti serotonínu.
Záver
Treba povedať, že endokrinné nádory – či už hormonálne aktívne alebo inaktívne – vyžadujú vždy multidisciplinárnu spoluprácu endokrinológa, internistu, chirurga, pracovníkov nukleárnej medicíny, genetika, onkológa a v neposlednom rade kvalitné laboratórium. Dôležitá je spolupráca a dispenzarizácia pacienta vedúca k vyliečeniu alebo aspoň stabilizácii základného ochorenia.
Literatúra:
- Kreze, A., Langer, P., Klimeš, I., Stárka, L., Payer, J., Michálek, J.: Všeobecná a klinická endokrinológa; 2004, 153 – 154
- Lazúrová, I., Payer, J. a kol.: Štandardné diagnostické a terapeutické postupy v endokrinológii, 2014, 63 – 69; 77 – 88; 162 – 166; 178 – 179; 19 2 – 194
- Ansell SM, Grant CS, Habermann TM. Primary thyroid lymphoma. Semin Oncol 1999; 26: 316 – 323
- Kiňová, S., Kekenáč, L., Kováčová, E., Koreň, M.: Multidisciplinárny prístup k liečbe gastroenteropankireatických neuroendokrinných tumorov. Vnitř. Lék.; 2010, 56(9); 946 – 950
- Kreze, A., Langer, P., Klimeš, I., Stárka L., Payer., J., Michálek, J.: Všeobecná a klinická endokrinológa; Hyperfunkcia kôry nadobličiek, 2004, s. 373 – 374
- Marek, J.: Doporučení hypofyzární společnosti po diagnostiku a léčbu prolaktinomu, Diabetologie a metabolizmus, endokrinologie, výživa., 2006; 9:149 – 155
- Podoba, J., Povinec, P.: Meniaca sa tvár primárnej hyperparatyreózy 1. časť. Etiopatogenéza, klinický obraz, diagnostika. Interná medicína 2013 (13), 62 – 69
- Podoba, J., Povinec, P.: Meniaca sa tvár primárnej hyperparatyreózy 2. časť. Indikácie liečby a terapeutické postupy. Interná medicíny 2013(13), 187 – 193




